











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Antecedentes
El síndrome de Wiskott-Aldrich (SWA) es un trastorno del sistema inmune ligado al cromosoma X que se comporta como una inmunodeficiencia combinada y se caracteriza por eccema, infecciones de repetición y trombocitopenia.1 Se debe a un defecto en la proteína WAS de 502 aminoácidos (cromosoma Xp 11.22-Xp 11.3.41), un regulador de la actina del citoesqueleto que se expresa en las células hematopoyéticas y está involucrada en la señalización celular, la migración y sinapsis inmunológica, debido a disfunción combinada de células T y B, con niveles de inmunoglobulina (Ig) G e IgM normales o bajos y elevación de IgA e IgE, lo que predispone a infecciones de repetición como otitis media recurrente, neumonía bacteriana y eccema atópico severo.
Otras manifestaciones del SWA son los trastornos autoinmunes, como anemia hemolítica autoinmune o púrpura trombocitopenia inmune (PTI), vasculitis de pequeños, medianos y grandes vasos, así como una alta susceptibilidad al desarrollo de tumores, debido a lo cual se han determinado diversos fenotipos, desde los que solo se manifiestan con trombocitopenia, hasta los que cumplen con todas las complicaciones de forma temprana o tardía.2,3
En 1937, Alfred Wiskott describió a tres hermanos con trombocitopenia congénita, melena, eccema y otitis media de repetición. Posteriormente, en 1954, Robert Aldrich identificó la herencia ligada al cromosoma X por la afectación de varios varones de una familia.4
Caso clínico
Niño de siete años, con PTI desde los tres meses de vida, quien presentó sangrado del tubo digestivo bajo y epistaxis, tratado con gammaglobulina intravenosa (GGIV) y bolos de metilprednisolona, 1 mg/kg/día de prednisona oral durante seis a 12 meses, con persistencia de sangrado tres o cuatro veces al año (melena, petequias y epistaxis), recuentos plaquetarios de 20 000 a 100 000/mm3. Al tratamiento se agregaron 3 mg/kg/día de azatioprina por dos años y globulina anti-D, sin mejoría, así como múltiples transfusiones de plaquetas, eritrocitos y plasma fresco durante los eventos agudos.
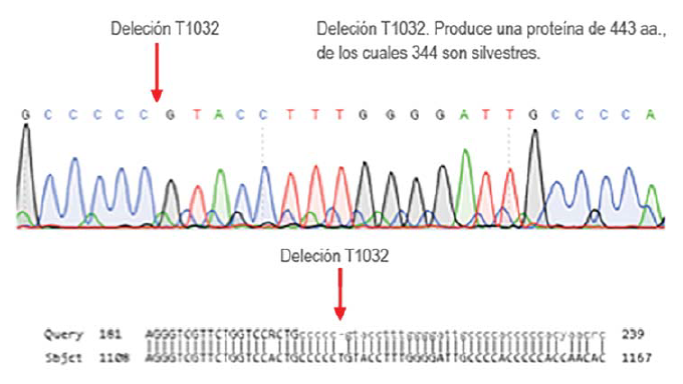
El paciente presentó otitis media supurativa en dos ocasiones y sinusitis maxilar bilateral. A los cinco años cursó con varicela complicada con choque séptico, por lo que ameritó intubación orotraqueal y antibióticos de amplio espectro (meropenem, vancomicina y aciclovir intravenoso). Se descartaron entidades infecciosas, con reevaluación de la patología autoinmune con autoanticuerpos negativos. Por la severidad de las infecciones se solicitó perfil inmunológico, con el que se identificó microtrombocitopenia y niveles bajos de IgG, CD4+ y CD19+ (Cuadro 1), por lo que se sospechó SWA, diagnóstico que se confirmó al demostrar mutación del gen WASP con deleción en T1032 en el Laboratorio de Inmunología Molecular de la Facultad de Medicina de Morelos, México (Figura 1).
Cuadro 1 Presentación clínica, estudios de laboratorio y gabinete en niño con síndrome de Wiskott-Aldrich quién presentó aneurisma aórtico
SWA = síndrome de Wiskott-Aldrich. GGIV = gammaglobulina intravenosa, TORCH = toxoplasmosis, rubéola, citomegalovirus, herpes.
Se dio tratamiento con 650 mg/kg de gammaglobulina intravenosa cada 21 días, profilaxis antimicrobiana con trimetoprima-sulfametoxazol y fluconazol, así como 0.7 mg/kg/día de prednisona y 2.5 mg/kg/día de ciclosporina debido a anemia hemolítica y uveítis. Se inició protocolo para trasplante de células progenitoras hematopoyéticas. Durante la evolución mostró mejoría de cuenta plaquetaria, con episodios de epistaxis leve y equimosis por traumatismos leves.
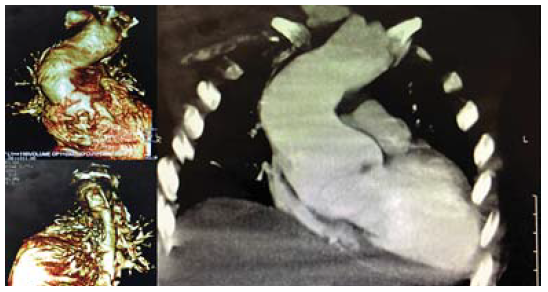
Una semana antes de la aplicación de la gammaglobulina intravenosa, el paciente fue llevado a urgencias por desviación súbita de la comisura labial izquierda; se encontró infarto en la cabeza del núcleo caudado y putamen del hemisferio izquierdo, sin datos de hipertensión intracraneal (Figura 2A). En la exploración física, el hallazgo fue soplo diastólico III/IV en foco aórtico; en la telerradiografía de tórax se observó cardiomegalia (Figura 2B) y en el ecocardiograma y la angiotomografía, dilatación aneurismática de la aorta ascendente (Figura 3). El paciente fue referido al servicio de cirugía vascular con clase funcional I y sin datos de bajo gasto cardiaco; al momento de este informe se encontraba en espera de la resolución quirúrgica y en tratamiento con propranolol, captopril y espironolactona, en tanto continuaba en protocolo de trasplante de células progenitoras hematopoyéticas como tratamiento de la IDP.
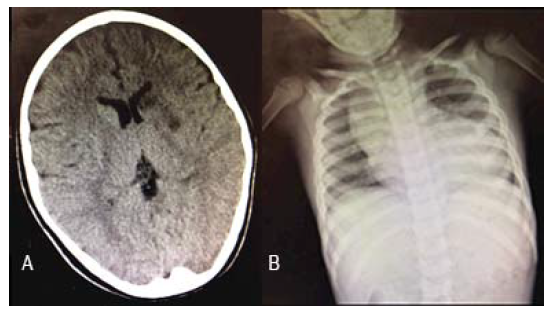
Figura 2 A) Tomografía axial computarizada simple de cráneo con imagen hipodensa en la cabeza del núcleo caudado y putamen del hemisferio izquierdo, así como discreto efecto de masa en el asta frontal ipsolateral, hallazgos compatibles con infarto isquémico. Atrofia cortical hacia la convexidad alta de los lóbulos frontoparietales. B) Radiografía de tórax con índice cardiotorácico de 0.67, ensanchamiento del mediastino a expensas de la aorta ascendente.
Discusión
El SWA es una inmunodeficiencia primaria ligada al cromosoma X, caracterizada por la asociación de microtrombocitopenia, eccema e infecciones de repetición debidas a deterioro variable de la inmunidad humoral y celular. Se han reportado desde fenotipos en los que solo existe compromiso plaquetario hasta los que presentan autoinmunidad o linfoma.2,3
La microtrombocitopenia se puede manifestar tempranamente con petequias, epistaxis, hematomas, sangrado de tubo digestivo y después de traumatismos o cirugías, debido a disminución del tamaño plaquetario, destrucción acelerada por defecto del citoesqueleto, secuestro plaquetario o anticuerpos antiplaquetarios.5
El eccema puede ser severo cuando existe el antecedente de atopia familiar, así como desequilibrio en la producción de la respuesta Th1 hacia Th2 con elevación de la IgE, lo que puede producir reacciones alérgicas. Las infecciones recurrentes pueden provocar una sinapsis inmune deficiente debido al defecto en la actina para la señalización celular, migración y maduración, con la consecuente función inmune deficiente, incluyendo células efectoras del sistema innato como fagocitos, monocitos y macrófagos, así como linfopenia generalizada, proliferación anormal de linfocitos T y poca respuesta a antígenos polisacáridos. En general, los niveles de la IgG son normales, los de la IgM están disminuidos y los de la IgA y IgE son normales o están incrementados.6
Los mecanismos de autoinmunidad en el SWA no son claros, pero se ha descrito producción defectuosa de células T reguladoras, con pérdida de la tolerancia central y periférica a autoantígenos, así como falla en la supresión de las respuestas inmunes excesivas.7
En el niño que describimos con PTI resistente al tratamiento convencional (esteroides e inmunosupresor) e infecciones recurrentes severas, se sospechó trastorno inmunológico. La determinación de microtrombocitopenia y linfopenia de CD4+ y CD19+ confirmó el diagnóstico de SWA al encontrar mutación del gen WASP. Sin antecedentes cardiovasculares previos, el paciente presentó infarto cerebral y soplo holodiastólico debido a dilatación aneurismática desde la raíz de la aorta hasta su porción descendente, con afección de la arteria coronaria.
Las anomalías cardiovasculares en el SWA son infrecuentes y debidas a vasculitis de pequeños, medianos y grandes vasos, incluyendo aneurisma de la aorta. Mediante estudios de patología se ha demostrado vasculitis necrosante. La patogenia de la vasculitis no está esclarecida, pero podría preceder a la microtrombocitopenia.8 Se describen reportes de casos con vasculitis y aortitis que se presentaron después de años del diagnóstico de SWA (Cuadro 2),incluso antes del trasplante de células progenitoras hematopoyéticas. La sintomatología cardiovascular se observó en menos de la mitad de los casos como disnea y opresión toráxica, mientras que en el resto el diagnóstico se realizó en forma fortuita con radiografía de tórax. Los pacientes tuvieron puntuación de 4 o 5 en la prueba clínica del síndrome de Wiskott-Aldrich. La resolución quirúrgica no se llevó a cabo de primera intención en todos y el trasplante de células progenitoras hematopoyéticas fue una opción inviable en algunos por falta de donador compatible.
Cuadro 2 Descripción de pacientes con SWA con aneurisma aórtico en distintos reportes de casos
| Autor y año | Diagnóstico SWA (años) | Descripción del aneurisma | Hallazgos para el diagnóstico | Aneurisma (años) | Otros hallazgos | Tratamiento | Pronóstico |
| Pellier et al. (20119) | ND | De aorta torácica descendiente | Radiografía de tórax | 10 | Progresión del aneurisma aórtico | TCPH | Vivo |
| ND | De aorta torácica | Radiografía de tórax | 13 | Insuficiencia valvular de la aorta, dilatación del ventrículo izquierdo | TCPH | Muerte por encefalitis viral posTCPH | |
| ND | De aorta abdominal | Ultrasonido abdominal | 13 | Dilatación de la arteria iliaca derecha | ND | Muerte por linfoma | |
| ND | De aorta torácica descendiente | Radiografía de tórax | 11 | No | ND | Vivo | |
| ND | De aorta ascendente | Dolor torácico | 16 | Disección aórtica, insuficiencia valvular | Cirugía, metotrexate, esteroides | ND | |
| Ono et al. (200910) | 5 | De aorta ascendente y aorta descendente | Radiografía de tórax | 7 | Leucemia linfocítica aguda, citomegalovirus | Cirugía y TCPH | Vivo |
| Onalan et al. (201811) | ND | De aorta descendente | Dolor torácico y disnea | 12 | Dilatación del ventrículo izquierdo | Cirugía | Vivo |
| Wood et al. (201712) | ND | De aorta descendente | Dolor torácico | 29 | Estenosis en el seguimiento | Dos cirugías | Vivo |
| Narayan et al. (200413) | 6 | De raíz aórtica y aorta ascendente | Disnea | 21 | Calcificación del aneurisma | Dos cirugías | Vivo |
| van Son et al. (199514) | ND | De aorta ascendente y aorta torácica descendente | Radiografía de tórax | 23 | Progresión de aneurisma aórtico e insuficiencia valvular | Cirugía | Vivo |
ND = información no disponible, TCPH = trasplante de células progenitoras hematopoyéticas.
Conclusión
La vasculitis puede presentarse en pacientes con inmunodeficiencias primarias que cursan con trastornos autoinmunes, como el SWA, en forma de aortitis con aneurisma aórtico, como en el paciente que describimos, lo que incrementa el riesgo de sangrado por la patología de base y el riesgo de ruptura del aneurisma. La evaluación cardiovascular en el SWA debería ser rutinaria, ya que la sintomatología puede estar ausente. Se debe priorizar el trasplante de células progenitoras hematopoyéticas como tratamiento definitivo.

