











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
Las simulaciones por computadora han desempeñado un papel fundamental al actuar como un puente que conecta la teoría con el experimento [1]. En algunos casos, estas simulaciones han adelantado predicciones experimentales [2] [3] o simplificado sistemas reales para abordar aspectos específicos que podrían oscurecerse en la complejidad del sistema en su conjunto [4]. En el presente artículo se profundizará en el ámbito de las simulaciones de dinámica molecular, destacando su papel crucial en el estudio de sistemas biológicos [5] [6] [7]. El enfoque principal de esta revisión recae en la membrana plasmática (MP), un componente esencial y complejo que actúa como manto protector de la célula. Las pequeñas variaciones en su entorno pueden desencadenar cambios significativos [8]. Comprender estas interacciones a nivel molecular es esencial para desentrañar los misterios de la biología celular.
El estudio de la estructura, función y los componentes de membranas es de suma importancia, por sus posibles aplicaciones para el diseño de nuevos fármacos en el tratamiento de enfermedades infecciosas, autoinmunes y degenerativas. No obstante, las propiedades biofísicas y el entendimiento de la termodinámica que influye en los estados de equilibrio de estas membranas son aún un tema abierto para la investigación [9] [10] [11].
Las simulaciones de dinámica molecular ofrecen una ventana única hacia el comportamiento molecular, superando las limitaciones de muchas técnicas experimentales actuales y proporcionando una visión detallada en escalas espacio-temporales, fundamentales para comprender los mecanismos que gobiernan la dinámica de la membrana [12]. A pesar de los avances en hardware y algoritmos, es necesario conocer los tiempos característicos del fenómeno que se quiere comprender.
Por ejemplo, los lípidos que componen una membrana plasmática pueden tener movimientos que duran desde picosegundos [13], pasando por nanosegundos, como la difusión de los lípidos en la membrana [14]; hasta horas, como el flip-flop [15]. Esta diversa gama de escalas temporales requiere distintos métodos para analizarlas debido a las limitaciones inherentes a cada método. La MP es un sistema complejo, lo cual obliga a estudiar sus componentes por separado. Esta aproximación permite comprender ciertos aspectos de la membrana sin las complicaciones de un sistema con diversas interacciones y funciones.
Esta revisión explorará la complejidad de las interacciones en la bicapa lipídica, destacando las dificultades inherentes al comportamiento de la membrana y cómo abordarlo. Se abordará la elección de escalas de simulación, desde la resolución detallada de todos los átomos [16], hasta la simplificación de grano grueso [17], y cómo esta elección afecta la precisión y el rendimiento computacional. Entender cómo ciertos comportamientos a nivel molecular dan paso a diversos fenómenos ha sido un reto desde que se tuvo conciencia de las implicaciones de las interacciones entre lípidos en una membrana.
Uno de los sistemas más estudiados y menos comprendidos es la MP, en concreto, los lípidos que la conforman y que, a diferencia de las proteínas y los genes, no revelan fácilmente cómo su estructura molecular causa alguna función biológica en la célula. Para entender el comportamiento de la MP, las simulaciones computacionales de sistemas modelo pueden darnos una visión del comportamiento a nivel molecular, inaccesible para la mayoría de las técnicas experimentales actuales, accediendo así a escalas espacio-temporales que nos ayuden a comprender los mecanismos que rigen la dinámica del sistema. Las simulaciones de dinámica molecular se pueden considerar un poderoso microscopio virtual, por lo que son una herramienta muy útil para estudiar membranas y otros sistemas biológicos.
A pesar de que los procedimientos de dinámica molecular son refinados continuamente para ser más veloces, precisos y aplicables a distintas circunstancias [18], aún se debe considerar el nivel de detalle que se pretende estudiar y los componentes de la membrana en los que se enfoca el análisis. En el caso de la MP, existen diversas escalas para modelar el sistema, como la escala de todos los átomos, la de átomo unido y la de grano grueso. En este trabajo, nos enfocaremos en los extremos: grano grueso y todos los átomos. La MP, además de actuar como manto protector de la célula, presenta complejos comportamientos termodinámicos, estructurales y dinámicos. A pesar de dicha complejidad, P. Van Der Ploeg logró en 1983 obtener información relevante a partir de un modelo muy simple de bicapa lipídica [19], el cual se componía de 16 moléculas de decanoato por monocapa y el sistema fue simulado por 80 ps. Aunque estos tiempos son pequeños, muchas de las herramientas de análisis de esa época siguen vigentes. El número de elementos en la bicapa de Van Der Ploeg y los tiempos de simulación no son comparables con los de una membrana natural ni con los tiempos de procesos biológicos, pero los resultados obtenidos sentaron las bases para estudios posteriores usando simulaciones computacionales. Esto confirma que podemos estudiar segmentos de la MP, como distintos tipos de proteínas o mezclas de lípidos, y así comprender aspectos de la membrana que resultarían oscurecidos por las numerosas interacciones presentes en la MP. Desde las simulaciones de Van Der Ploeg, ha habido enormes avances en los potenciales modelos (force fields), en las técnicas, algoritmos de simulación y en las capacidades de procesamiento numérico masivo. Sin embargo, aún existen limitaciones para simular sistemas con el número de componentes de una célula y los tiempos de simulación requeridos para estudiar dichos fenómenos [20].
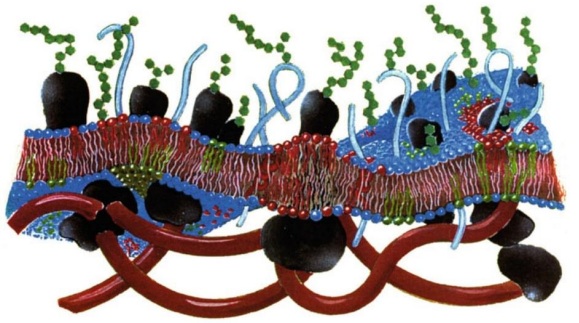
Figura 1: Representación esquemática de la membrana plasmática. Con la bicapa como estructura central, la cual presenta ondulaciones, segmentos con un mayor espesor de bicapa y heterogeneidad lateral. También se observan algunas proteínas en color negro y el citoesqueleto de color rojo en el exterior de la bicapa. [22]
Dependiendo de lo que se quiera analizar en la simulación, se pueden usar distintas escalas. Las simulaciones en escala de todos los átomos incluyen todos los enlaces químicos y todas las interacciones posibles entre los átomos, pero son computacionalmente caras y pueden ser imprácticas para sistemas grandes o largos tiempos de simulación. En la resolución de todos los átomos, se deben resolver las ecuaciones de movimiento de Newton para cada átomo, repitiendo estos cálculos millones de veces, lo que implica un gran tiempo de espera para obtener resultados.
En el caso de grano grueso, se proponen pseudo átomos compuestos por varios átomos, lo que permite simulaciones más largas y con más componentes, pero sacrificando precisión. La decisión de utilizar una escala u otra depende del enfoque del trabajo y lo que se pretenda investigar. Las simulaciones de grano grueso pueden incluir más elementos y obtener simulaciones más largas por el mismo costo computacional que un sistema más pequeño y con menor tiempo de simulación en escala de todos los átomos. Sin embargo, las interacciones lípido-lípido son muy diversas y cualquier simplificación tiene consecuencias. Ahondaremos más en el tema en las siguientes secciones.
A continuación, hablaremos de la MP en su conjunto, la bicapa como un modelo para entender ciertos mecanismos que ocurren en la membrana y las características especiales de esta. Después, daremos una breve introducción a las simulaciones de dinámica molecular, los retos de estudiar bicapas usando este método y las escalas más usadas en el análisis de bicapas. Por último, presentaremos las conclusiones.
Membrana plasmática: explorando la complejidad celular
La MP es la capa protectora de la célula, la cual se erige como un complejo entramado de elementos esenciales. Algunos de ellos son la bicapa lipídica, como elemento central; proteínas con diversas funciones y configuraciones, y el citoesqueleto, que le confiere propiedades mecánicas a la membrana. Muchos procesos biológicos de importancia en la célula, ocurren o son mediados por la bicapa, como el transporte, la función neuronal, la respuesta inmunológica, entre otros, por lo que la comprensión detallada de estos componentes se convierte en un imperativo para desentrañar los misterios de la MP en su totalidad.
Ante la vasta diversidad de moléculas y los diversos tiempos característicos de los fenómenos en la MP, surge la necesidad de abordar el estudio de sus componentes de manera individual. Esta estrategia, aunque restringe variables, ofrece un entendimiento detallado de cada componente, permitiéndonos responder algunas preguntas de las muchas que existen.
Estudiar una membrana con todos sus componentes es imposible con las técnicas experimentales actuales. Por esta razón los investigadores suelen centrarse en aspectos específicos de la MP. Esta aproximación más enfocada, permite abordar de manera más efectiva la complejidad del sistema. Por ejemplo, podríamos centrarnos exclusivamente en la bicapa lipídica, dejando de lado los demás elementos, lo que permite analizar las interacciones entre lípidos y sus efectos sin necesidad de considerar otros aspectos de la MP.
La bicapa, componente esencial de la célula, actúa como el epicentro de la MP. Su estructura, determinada por las especies de lípidos que la componen, puede sufrir cambios drásticos al alterar la concentración o al introducir una especie diferente [21]. Estos comportamientos revelan que interacciones aparentemente simples pueden tener efectos profundos en el comportamiento global de la bicapa.
Fases de la Bicapa
Para caracterizar adecuadamente a nuestro sistema es importante entender las diferencias estructurales de la bicapa en sus distintas fases y, a su vez, qué motiva los cambios dinámicos y estructurales en los lípidos como unidad y en su conjunto. En general, cuando se habla de diferentes fases en la materia, nos referimos a distintos grados de orden. Un sólido se considera ordenado cuando mantiene una estructura regular, como se muestra en la Figura 2(a), a diferencia de la Figura 2(c) que es una fase líquida y no tiene un orden aparente.
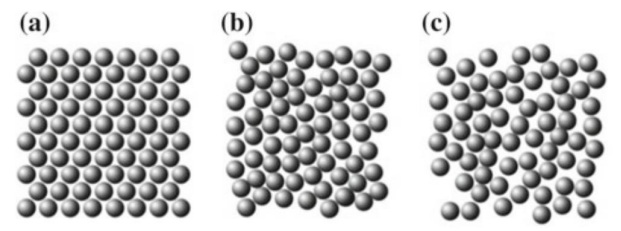
Figura 2: Descripción pictórica de distintas fases posibles en un sistema compuesto sólo por esferas que no se deforman. (a) representa un sólido, (b) un sólido amorfo o un líquido muy viscoso y (c) representa a un líquido ordinario.
Pensemos en la representación pictórica de la figura 2. En este caso, los componentes del sistema son esferas perfectas. La simetría esférica de estos elementos impide que se observe algún cambio al rotar cualquiera de ellos. Es decir, para esta clase de sistemas sólo existe orden espacial, ya que al rotar los elementos no se observa ningún cambio.
Sin embargo, para una aproximación más adecuada en el caso de lípidos, debemos considerar estructuras con forma esferoide. Así, no sólo existe un orden o desorden de los componentes en el espacio, sino también uno asociado a los ejes de simetría del lípido. En este contexto, el término “orden” también se refiere a la forma en la que están orientados los elementos del sistema.
Considerando componentes con geometrías como las mencionadas en el párrafo anterior, las fases pueden ser más diversas, dando paso a lo que se conoce como meso fases. En la figura 3(a) se observa un orden tanto espacial como orientacional propio de un sólido; en la figura 3(b) se presenta una mesofase en la que se tiene un cierto grado de orden, ya que los elementos están localizados en planos paralelos con una distancia fija, como en un cristal y con los componentes orientados en la misma dirección. Sin embargo, en cada plano se tiene desorden espacial como en un líquido bidimensional. A esta mesofase se le llama cristal líquido esméctico y una bicapa lipídica es un ejemplo de este tipo de fase en particular [22]. Por último, en la figura 3(c) observamos el análogo de una fase líquida donde no existe orden en el espacio ni en la orientación.

Figura 3: Descripción pictórica de meso fases en un sistema con componentes que tienen forma esferoide.
Cuando se estudia un sistema como la bicapa y hablamos de cambio de fase, es esencial considerar la transición de fase principal, que se cumple para todos los lípidos. Esta transición ocurre al pasar de la fase de sólido ordenado a la fase de líquido desordenado.
La transición principal está asociada a una fusión de las cadenas hidrocarbonadas en el sentido en que, cuando la temperatura es menor a la temperatura de transición (
Además de estas dos fases intrínsecas a todos los lípidos, en diversas publicaciones [23] [24] [25] se habla de un proceso de reorganización de lípidos, fenómeno al que se le ha adjudicado un importante papel en distintas tareas celulares, como la transducción de señales o su rol como plataforma para el acceso de virus. Con esto nos referimos a la hipótesis de las balsas lipídicas [26].
Se cree que las balsas se forman por la manera en la que interactúa el colesterol con distintos tipos de lípidos. Al estar el colesterol presente en un sistema en el que coexisten lípidos en fase de
La fase
La estructura del sistema depende del contexto lipídico
Diversos factores pueden cambiar la estructura del sistema, por ejemplo, la diferencia de tamaño entre lípidos aledaños obligaría a los componentes del mismo tamaño a alinearse entre sí, produciendo dominios con distintos espesores de membrana. Otro factor capaz de cambiar la estructura del sistema es la temperatura; como en cualquier sistema termodinámico, la temperatura guarda una estrecha relación con la energía cinética de los componentes del sistema, por lo que reducir la temperatura generaría una membrana más rígida y en la que las cadenas hidrocarbonadas de sus componentes estarían más extendidas y, en el caso contrario, una estructura más fluida, con las cadenas con una mayor movilidad.
Es también importante tomar en cuenta el grado de insaturación de las cadenas hidrocarbonadas de los lípidos en el sistema, ya que dependiendo de si son lípidos saturados, insaturados, mixtos e, incluso, dependiendo del lugar geométrico donde se encuentre el doble enlace en la cadena, se pueden tener dominios con mayor o menor fluidez. En general, los lípidos saturados son más rígidos y tienden a tener interacciones más cercanas entre ellos, originando regiones con baja fluidez; por otro lado, los insaturados no permiten una relación tan estrecha, por lo que dan lugar a regiones con mayor fluidez. Y, por último, pero no menos importante, se debe tener en cuenta el efecto regulador que el colesterol tiene al encontrarse a distintas concentraciones en una membrana.
Queda claro que los componentes que se escojan para modelar la membrana determinarán, al final de cuentas, las estructuras que se formen en ella, por lo que todos los factores antes mencionados deben tomarse en cuenta. Como ejemplo de las variaciones en el comportamiento de una membrana al cambiar uno de los componentes, podemos observar la Figura 5. En este caso en particular, sólo el tamaño de las cadenas de uno de los lípidos en un sistema binario fue modificado y esto llevó a cambios significativos en el tipo de fases que se forman en el sistema. En este caso, tenemos temperaturas en el eje Y y concentraciones de DPPC en el eje X, f es fase de líquido desordenado y g es fase sólido ordenado. Lo que sugiere Risbo y colaboradores (1995) [21] es que, conforme el tamaño de las cadenas de uno de los lípidos disminuye, crece la zona de coexistencia de fases entre sólido ordenado y líquido desordenado, hasta llegar a formar nuevas fases de sólido ordenado con distintas características (
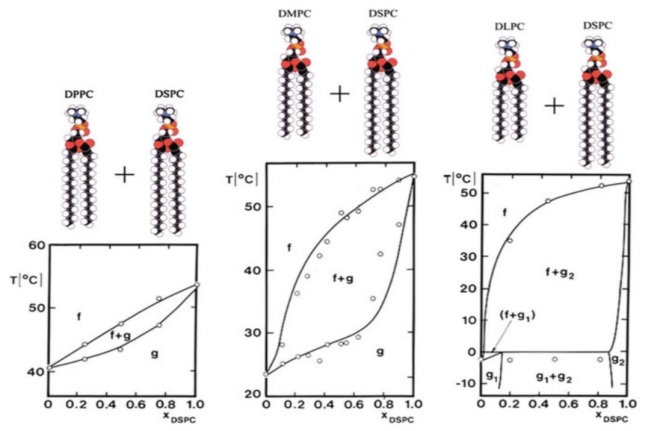
Figura 5: Diagrama de fase de mezcla binaria de tres sistemas. De derecha a izquierda se reemplaza uno de los lípidos por uno con cadenas más pequeñas. Figura tomada de referencia [21].
Simulaciones de dinámica molecular
Actualmente, la velocidad de cálculo y procesamiento de las computadoras ha permitido el desarrollo de diferentes técnicas y algoritmos de simulación, los cuales han sido ampliamente utilizados para estudiar sistemas biológicos y los diversos procesos que ocurren en ellos a nivel
molecular La dinámica molecular es una técnica bien establecida y su uso es cada vez más común debido a su gran éxito en replicar resultados experimentales; sin embargo, la confiabilidad de la imagen molecular que se obtenga del cálculo depende en gran medida de la correcta representación de las interacciones moleculares, asociadas al campo de fuerza que se utilice. En la dinámica molecular se resuelven de manera numérica las ecuaciones de movimiento de Newton para un conjunto N de átomos con coordenadas cartesianas
En este trabajo no ahondaremos en los detalles de cómo son descritas las interacciones entre moléculas en un sistema biológico, para un mayor detalle se puede recurrir al artículo “Métodos de simulación computacional en biología” [27]
Para analizar con dinámica molecular sistemas biológicos, debemos considerar un ensamble que nos ayude a replicar datos experimentales, por lo que el ensamble NPT es idóneo [28] [29]. En este ensamble, la presión, temperatura y número de moléculas se mantienen constantes, pero es claro para nosotros que en las ecuaciones de movimiento de Newton no existen parámetros que incluyan a la temperatura ni a la presión, por lo que las ecuaciones deben ser modificadas para incluirlos [30] [31].
Campo de fuerza
Un campo de fuerza [32] es una expresión matemática que describe la dependencia de la energía con las coordenadas de las partículas en el sistema. Para desarrollar un campo de fuerza es necesario un potencial V(R), donde R es un vector de posición generalizado como se muestra en la siguiente ecuación.
Los primeros tres términos representan interacciones entre átomos de una misma molécula y el cuarto y quinto término representan interacciones entre elementos de distintas moléculas. Los parámetros que aparecen en la ecuación (2) son aquellos que resultan adecuados para reproducir resultados experimentales.
Dificultades en el estudio de membranas modelo
Como ya se ha mencionado, debido a la complejidad del sistema en la naturaleza, se deben hacer ciertas simplificaciones al hacer simulaciones. Por un lado, se utilizan campos de fuerza que deben ser capaces de reproducir propiedades experimentales del sistema que se quiere modelar [32] [33] y por otro lado se deben tener en cuenta las limitaciones de dicho modelo y decidir si estas limitaciones afectan a lo que pretendemos medir.
Otra dificultad radica en el número de lípidos que se pueden incluir en una simulación de membrana a diferencia de una en la naturaleza, ya que la capacidad de cómputo que se tiene debe ser aprovechada de manera que se puedan obtener conclusiones en tiempos relativamente cortos. De manera similar, el tamaño de la celda de simulación puede llegar a ser una limitante, así como los distintos tipos de lípidos que se pueden incluir en la simulación, ya que podría no haber suficientes datos experimentales para poder modelar sus cualidades específicas.
Escala de todos los átomos y de grano grueso
Debido a la complejidad de las distintas escalas que se presentan en el estudio de membranas y ya que es posible estudiar una gran diversidad de fenómenos que pueden ocurrir en escalas muy distintas de tiempo ―por ejemplo, la rotación o movimiento de las cadenas hidrocarbonadas puede medirse en escalas de decenas de picosegundos― la traslación de fosfolípidos en la membrana se encuentra en el rango de nanosegundos y procesos como el flip-flop pueden tomar horas, quizá días. Tomando en cuenta que las simulaciones de dinámica molecular de membranas calculan posiciones cada par de femtosegundos y si se requieren simular, al menos, tiempos de microsegundos, esto conlleva un gran costo computacional; no se diga simular horas o días.
Por lo tanto, es importante decidir el nivel de detalle a estudiar; para ello, se tienen dos representaciones que son muy socorridas, la de todos los átomos y la de grano grueso que podemos ver ejemplificadas en la Figura 6. En el primer caso, como su nombre lo dice, se consideran todos los átomos que constituyen a cada fosfolípido en el sistema, en dinámica molecular, esta es la representación más precisa pero la que tiene mayores limitantes al decidir el tamaño de la celda de simulación, número de fosfolípidos e, incluso, el número de replicas que se pueden hacer del sistema. Sin embargo, si lo que se quiere estudiar es, por ejemplo, los puentes de hidrógeno entre distintos elementos en la MP, es necesario utilizar este tipo de representación para poder observar estas interacciones. Por otro lado, en la naturaleza tenemos todos los átomos interactuando en mayor o menor medida, por lo que esta representación es considerada la que da resultados más fidedignos. También tenemos la descripción de grano grueso en la que se generan seudo átomos a partir de la representación de todos los átomos. En el modelo de MARTINI, 4 átomos se encasillan en un seudo átomo tal que se tienen un menor número de átomos a considerar y podemos tener simulaciones por tiempos más largos y con un mayor número de lípidos, pero perdiendo todo el detalle atomístico.
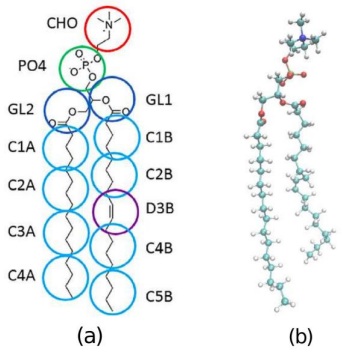
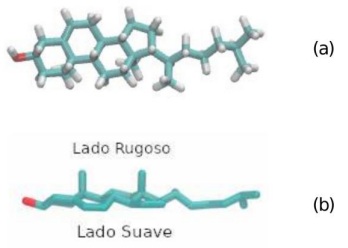
Figura 6: Representación en grano grueso (a) y representación en escala de todos los átomos en (b) del mismo fosfolípido.
Por ejemplo, consideremos la hipótesis de las balsas lipídicas, discutida previamente. En esta teoría, se cree que el colesterol actúa como un agente reorganizador de los lípidos, dependiendo de su afinidad con ellos. No es absurdo pensar que una bicapa en escala de grano grueso podría darnos una buena aproximación de la organización lateral de sus componentes y ahorrarnos tiempo de cómputo que podría verse cuadruplicado en el caso de escala de todos los átomos. Sin embargo, al reflexionar respecto a la hipótesis, recordamos que la formación de fases de
Otro detalle importante que considerar al utilizar grano grueso al estudiar la hipótesis de las balsas lipídicas es la redefinición de la estructura del colesterol al pasar de todos los átomos a grano grueso. En la mayoría de los modelos el lado rugoso y el lado suave terminan siendo muy similares, lo cual puede ser un problema, ya que son las diferencias entre estos dos lados lo que, se cree, le da cualidades al colesterol para actuar como mediador entre dos lípidos.
Tomando en cuenta el estudio de Ingólfsson [7] en el que se incluyen más de 60 tipos de lípidos en una membrana muy grande, con tiempos de simulación de decenas de microsegundos, podemos entender las bondades del modelo de grano grueso, ya que, en el caso de todos los átomos, incluso una mezcla de tres componentes puede llegar a ser un reto y simular decenas de microsegundos podría tomar años en un GPU de relativamente buena calidad. En este sentido, el utilizar grano grueso nos puede dar una ventaja, ya que podemos modelar interacciones entre muchos tipos de distintos lípidos como ocurre en la naturaleza. De manera general ella Tabla 1 se presentan las ventajas y desventajas de usar distintas escalas en simulaciones de dinámica molecular.
Tabla 1: Ventajas y desventajas generales para distintas escalas en simulaciones de dinámica molecular.
| Escala de la simulación | Pros Notables | Contras Notables |
| Todos los átomos | Se considera el detalle químico completo de los lípidos, ajustado y optimizado de acuerdo con diversas medidas experimentales. | Es caro computacionalmente y, por esa razón, impráctico en el estudio de membranas de gran tamaño o que requieren tiempos largos de simulación. |
| Átomo unido | Es detallado, pero sin incluir los átomos de hidrógeno, debido a esa simplificación puede ser ~2-3 veces más eficiente que la escala de todos los átomos. | Sigue siendo caro computacionalmente y resulta impráctico en el estudio de membranas de gran tamaño o que requieren tiempos largos de simulación. |
| Grano grueso | Es al menos un orden de magnitud más eficiente que todos los átomos y átomo unido y, por lo tanto, puede acceder a tiempos más largos de simulación y sistemas más grandes. | Es menos preciso y solo permite estimaciones aproximadas o descripciones generales. Este método a veces altera la dinámica natural del sistema, ya que simplifica la complejidad energética del paisaje de la bicapa. |
Conclusiones
En el estudio de simulaciones de membrana modelo, se deben tomar en cuenta qué elementos van a ser incluidos en el sistema, ya que las diferentes clases de lípidos producen distintas interacciones y, dependiendo del proceso biológico que se requiera modelar, algunas mezclas pueden dar mejores resultados que otras. También existen procesos que se desarrollan a diversas escalas temporales y ciertos detalles pueden no ser tomados en cuenta por el modelo si elegimos una escala que no toma en cuenta las interacciones intrínsecas al fenómeno a estudiar. Por un lado, las simulaciones de todos los átomos son capaces de describir con total detalle atomístico una membrana, con las limitaciones antes mencionadas, pero sin perder de vista que los sistemas en la naturaleza no discriminan ciertas interacciones por ser “costosos computacionalmente” y, por otro lado, tenemos la escala de grano grueso que nos permite darnos una idea de sistemas más complejos en los que se incluyen una gran cantidad de constituyentes por tiempos de simulación inimaginables, a la fecha, para sistemas en escala de todos los átomos.

