











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkEl cerdo pelón mexicano (CPM) es una subpoblación de cerdos criollos1, su origen se remonta a la colonización del continente americano. En Europa, las bases genéticas del cerdo se atribuyen a cuatro líneas: Céltica, Ibérica, Napolitana y Asiática2,3. El arraigo y distribución a través del continente americano tuvo trascendencia por la interacción genotipo -medio ambiente4; los efectos de selección, regulada por los humanos y el ambiente, han generado distintas razas y líneas con sus particularidades productivas y funcionales5,6. El CPM es un reservorio de material genético3, con alta rusticidad, adaptación a climas adversos y resistencia a enfermedades, se desplaza por largas distancias en terrenos hostiles y con escasez de alimento7,8,9; además, presenta carne de buena calidad debido a la grasa intramuscular que deposita10.
Los patrones de variación genética brindan información sobre mecanismos genéticos asociados a rasgos de adaptación y producción. La expresión genética realiza un papel importante durante el transcurso de domesticación y evolución, descifrar los mecanismos de selección subyacentes no solo beneficia la conservación y crianza, sino que también impacta en la identificación de genes asociados con procesos biológicos, rasgos productivos y reproductivos de interés11. Los procesos de selección dejan huella en el genoma, tales como reducción de la diversidad genética y la existencia de haplotipos12. Aplicaciones de genómica, a partir de secuenciación de alta densidad, han hecho posible la identificación de señales de selección, caracterizando sucesos de selección natural y artificial5,13. Las señales de selección (SS), se asumen con la detección de regiones genómicas en las que se ubican variantes genéticas expuestas a un aumento rápido de frecuencias alélicas, dado la presión de selección, o la determinación de regiones genómicas fijadas en una población con fenotipos establecidos5.
La estadística basada en haplotipos detecta un rápido incremento de un alelo seleccionado a una frecuencia media, en el transcurso del cual, el rango de asociación de haplotipos no se pierde por recombinación14. La exploración en el genoma en búsqueda de SS ha detectado señales de genes candidatos asociados a variables fenotípicas y rasgos de interés económico15. Dado la relevancia del CPM, como recurso genético con cualidades de adaptación y rusticidad, así como potenciales productivos, los objetivos del presente estudio fueron identificar la posibles SS, así como como asociar genes candidatos (GC) y loci de caracteres cuantitativos (QTL) con las regiones que denoten selección.
Se analizaron los genotipos de 107 CPM, muestreados en 12 unidades de producción, bajo el esquema de traspatio y extensivo, ubicadas en los estados de Yucatán, Campeche y Quintana Roo. Los cerdos muestreados cumplen las especificaciones del patrón racial del CPM y derivan del registro genealógico de la Asociación Mexicana Especializada en Cerdo criollo16. Los genotipos conforman el panel de 50657 SNP (Polimorfismos de Nucleótido Simple) integrados en el chip GGP Porcine BeadChip 50K. Se descartó los SNP con frecuencia de alelo menor (MAF) por debajo de 0.05 y tasa de genotipado por debajo de 0.90, así como individuos con más de 10 % de genotipos faltantes. Posterior a la edición, los genotipos faltantes fueron imputados, formando haplotipos por desequilibrio de ligamiento con el software SHAPEIT v.217. Con la librería CMplot18 de R se generó el gráfico de posición de SNP en el genoma porcino Sus scrofa 10.2, según su ubicación a través de cromosomas. Para la detección de SS se utilizó el método de integración de haplotipos19 (iHS; integrated haplotype score), el cual es una variante del método de homocigosidad en haplotipos (EHH; haplotype homozygosity)20. El iHS requiere la presencia del alelo ancestral, identifica selecciones positivas, dado que la frecuencia del haplotipo derivados se ve incrementada. El análisis se realizó con la librería rehh 2.0 del paquete R21, el estadístico de prueba se generó a partir de la ecuación:
Donde: iHS(s), puntuación de haplotipos integrados; iHHa, área bajo la curva de la extensión de la homocigosidad haplotípica para los alelos ancestrales; iHHd, área bajo la curva de la extensión de la homocigosidad haplotípica para los alelos derivados; µps, media de la estandarización, calculada sobre todos los SNP con frecuencia alélica derivada (ps) similar a la del núcleo SNP (s); σps, desviación estándar de la estandarización, calculada sobre todos los SNP con frecuencia alélica derivada (ps) similar a la del núcleo SNP (s). Posterior a la estandarización de los datos se realizó la transformación piHS con base en la ecuación piHS = − log10 (1 − 2|Φ (iHS) − 0.5|); donde: Φ (x) representa la función de distribución acumulativa gaussiana.
Los SNP tienen una aproximación a la distribución neutral y piHS se interpreta como un valor de probabilidad (Pval; en una escala - log10) asociado a la hipótesis neutral de no selección, los marcadores que expresaron un Pval<0.0001 se tomaron como significativos que presentan señal de selección. Se utilizaron los alelos ancestrales derivados del estudio realizado por Bianco et al22, donde reportan un catálogo de variaciones autosómicas de un solo nucleótido utilizando cuatro especies de Sus (Sus barbatus, Sus celebensis, Sus verrucosus y Sus cebifrons) y una especie de Phacochoerus (warthog africano) como grupo externo. Con la librería pegas del paquete R23 se realizó la gráfica de los haplotipos derivados del desequilibrio de ligamiento de los cromosomas con mayor número de marcadores con significancia ante la señal de selección. Con los marcadores con SS significativa (pval<0.0001) se realizó la asociación de GC y QTL mediante la alineación de posiciones utilizando la librería Genomic Ranges del paquete R24, el cual se basa en asignar cada SNP a todos los genes localizados parcial o completamente en una región de 0.75 Mb alrededor de cada loci, a partir de las referencias publicadas en el NCBI (National Center for Biotechnology Information) y el EBI (European Bioinformatics Institute). Se utilizó la librería BiomaRt del paquete R25 para acceder a la base de datos Ensembl y realizar el traslape de genes candidatos con las regiones que denotaron SS. La información de los QTL fue consultada en la base de datos Pig QTLdb26.
Posterior al proceso de edición se estimó el iHS para 33499 SNP. En la Figura 1 se presentan los marcadores o regiones genómicas que presentan señal de selección significativa (P<0.0001), así como los iHS y piHS distribuidos a través de los diversos cromosomas. El resultado mostró disminución de homocigosidad haplotipica sobre un loci, con base en la comparación entre el alelo derivado y el ancestral dentro de una población determinada; valores extremos de iHS (negativos o positivos) denotan una homocigosidad haplotipica excesiva sobre los alelos ancestrales o derivados, de tal manera que las regiones genómicas que presentan picos de iHS en una o ambas direcciones son importantes debido a que han sufrido selección.
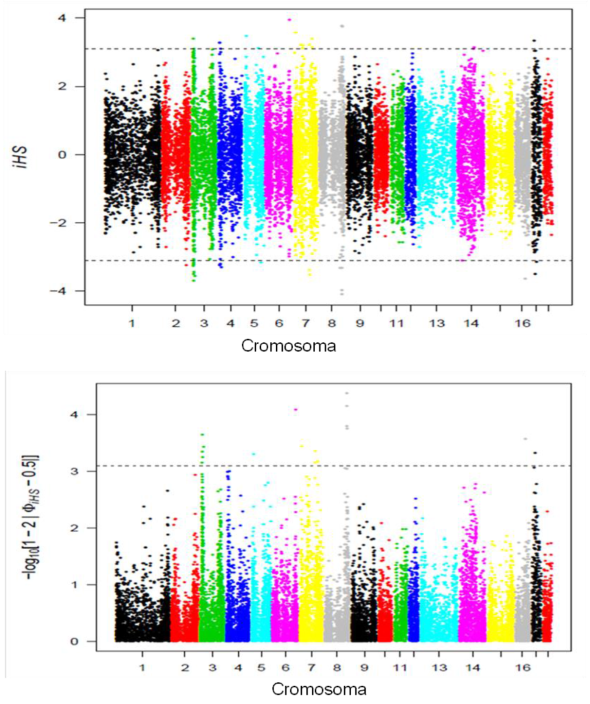
Los valores que rebasan la línea punteada denotan significancia estadística (P<0.0001).
Figura 1 Gráficos de tipo Manhattan con valores iHS halotipos (integrated haplotype score), halotipos y la transformación piHS (piHS = − log10 (1 − 2|Φ (iHS) − 0.5|)
Fueron 20 marcadores los que denotaron significancia (P<0.0001) a la SS (Cuadro 1), distribuidos en los cromosomas 3, 5, 6, 7, 8, 16 y 17.
Cuadro 1 Marcadores SNP con significancia estadística (P<0.0001) en las regiones genómicas con señales de selección
| SNP | CHR | Posición (pb) | iHS | piHS | P-valor |
|---|---|---|---|---|---|
| WU_10.2_8_128404843 | 8 | 128404843 | -4.094 | 4.373 | 4.24E-05 |
| WU_10.2_8_128418568 | 8 | 128418568 | -3.976 | 4.154 | 7.01E-05 |
| ASGA0096926 | 6 | 134750885 | 3.941 | 4.090 | 8.13E-05 |
| WU_10.2_8_128447508 | 8 | 128447508 | 3.776 | 3.797 | 1.59E-04 |
| WU_10.2_8_128761047 | 8 | 128761047 | 3.753 | 3.757 | 1.75E-04 |
| WU_10.2_3_14556426 | 3 | 14556426 | -3.690 | 3.649 | 2.25E-04 |
| WU_10.2_16_59651156 | 16 | 59651156 | -3.645 | 3.574 | 2.67E-04 |
| WU_10.2_7_12543606 | 7 | 12543606 | 3.568 | 3.445 | 3.59E-04 |
| MARC0081434 | 3 | 21071325 | -3.563 | 3.436 | 3.66E-04 |
| WU_10.2_7_88718236 | 7 | 88718236 | -3.514 | 3.355 | 4.42E-04 |
| ASGA0013651 | 3 | 15463031 | -3.509 | 3.347 | 4.50E-04 |
| WU_10.2_17_27206748 | 17 | 27206748 | -3.493 | 3.320 | 4.78E-04 |
| WU_10.2_5_10602026 | 5 | 10602026 | 3.483 | 3.304 | 4.96E-04 |
| WU_10.2_3_16123577 | 3 | 16123577 | -3.446 | 3.245 | 5.69E-04 |
| MARC0106494 | 7 | 101800928 | 3.405 | 3.179 | 6.63E-04 |
| WU_10.2_3_13551908 | 3 | 13551908 | 3.390 | 3.155 | 7.00E-04 |
| ALGA0042950 | 7 | 87847151 | -3.385 | 3.148 | 7.12E-04 |
| ASGA0075836 | 17 | 21585923 | 3.332 | 3.064 | 8.62E-04 |
| WU_10.2_8_118363092 | 8 | 118363092 | -3.327 | 3.056 | 8.79E-04 |
| WU_10.2_8_128287306 | 8 | 128287306 | -3.319 | 3.044 | 9.04E-04 |
SNP= nombre de referencia del SNP; CHR= cromosoma de Sus scrofa; Posición (pb) = pares de bases; iHS= puntaje del haplotipo; piHS= puntaje del haplotipo transformado.
Los haplotipos de los cromosomas con presencia de marcadores con selección se muestran en la Figura 2; el cromosoma 8 presentó mayor conservación e intensidad, con posible asociación a respuesta a la selección en recientes generaciones; en los cromosomas 3 y 7 se observaron haplotipos pequeños y menos delimitados, como indicador de selección natural o adaptación.
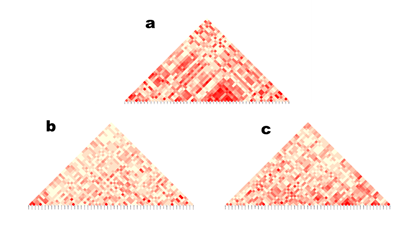
El eje de abscisas representa los loci donde se está midiendo el desequilibrio de ligamiento; el eje vertical representa los loci de contraste con el que se midió el desequilibrio de ligamiento. El valor dentro de cada celda (mapa de calor; intensidad de color) representa el valor del desequilibrio de ligamiento (r²= 0 a 1) entre los loci correspondientes; a mayor intensidad en el color, muestra desequilibrio (los alelos en esos loci se heredarán juntos); baja intensidad, indican que los loci son independientes.
Figura 2 Haplotipos de los cromosomas significativos: a) cromosoma 8; b) cromosoma 7; c) cromosoma 3
En la Figura 3 se muestra la estructura de haplotipos alrededor del alelo central (WU_10.2_8_128404843) con mayor piHS (P<0.00005). El diagrama bidireccional permite interpretar el origen de una SS, traza el desglose de DL a distancias cada vez mayores del alelo central en el SNP focal seleccionado. La raíz (SNP focal) del diagrama es el alelo central y se identifica mediante una línea punteada de forma vertical. El diagrama es bidireccional y representa la DL centrómero proximal y el centrómero distal. En una dirección, cada marcador es una oportunidad para un nodo, el diagrama se divide o no (en función de si ambos o solo un alelo) está presente; el desglose de DL en el fondo del haplotipo central se representa a distancias progresivamente más largas. El grosor de las líneas corresponde al número de individuos con el haplotipo de larga distancia indicado, a mayor número de individuos, el grosor será mayor21.
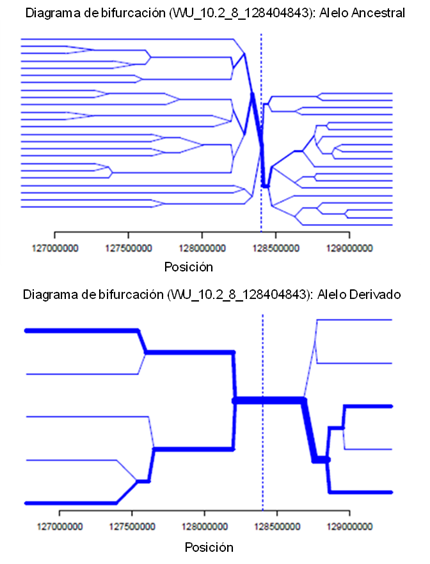
Figura 3 Estructura de haplotipos (posición en pares de bases) alrededor del alelo central (WU_10.2_8_128404843)
En las regiones candidatas que presentaron selección se identificaron genes ENSEMBL a través de cinco cromosomas (Cuadro 2). Las regiones con SS fueron: 15026769 a 103492964 del cromosoma 3; 10553433 a 66661654 del cromosoma 5; 134659633 a 134777072 del cromosoma 6; 12300999 a 102777822 del cromosoma 7; y, 118251236 a 128953331 del cromosoma 8. En la alineación de QTL, se asociaron 146 distribuidos en 7 cromosomas, ubicados en las regiones: 1456046 a 143758669 del cromosoma 3; 844337 a 109562487 del cromosoma 5; 25859176 a 146365886 del cromosoma 6; 48748 a 134608551 del cromosoma 7; 3470575 a 148491826 del cromosoma 8; 5836383 a 209900460 del cromosoma 13; y, 342954 a 83197113 del cromosoma 16. Los QTL identificados se agrupan en: comportamiento (tiempo dedicado a alimentarse, comportamiento de afrontamiento); conformación (posición del oído y área de la oreja, número de vértebras, conformación de patas y piernas); gordura (área, peso, porcentaje y distribución de grasa, marmoleo, diámetro de los adipocitos); capacidad inmune (nivel del receptor tipo toll 2 y 9, proliferación celular inducida, concentración de C3C, porcentaje de leucocitos positivos, actividad hemolítica del complemento, número de neutrófilos segmentados, recuento de glóbulos blancos, actividad fagocítica); reproducción (capacidad uterina, peso del tracto reproductivo, número de pezones, edad en la pubertad, duración de la gestación).
Cuadro 2 Genes ubicados en regiones con señal de selección
| Nombre del gen | ID | Función | Dominio |
|---|---|---|---|
| ENSSSCG00000007733 | GO:0005509 | Unión de iones de calcio | Función molecular |
| GALNT17* | GO:0000043 | Polipéptido N-acetil galactosaminil transferasa |
Función molecular |
| VIT* | GO:0000107 | Vitrin | Procesos biológicos |
| RAC2 | GO:0007264 | Pequeña transducción de señales mediada por GTPasa |
Procesos biológicos |
| SSTR3 | GO:0004994 | Actividad del receptor de Somatostatina |
Función molecular |
| C1QTNF6 | GO:0005515 | Enlace proteico | Función molecular |
| ENSSSCG00000029668 (IL2RB) |
GO:0004896 | Actividad del receptor de citocinas |
Función molecular |
| TMPRSS6 | GO:0006508 | Proteólisis | Procesos biológicos |
| CRACR2A | GO:0003924 | Actividad de GTPasa | Función molecular |
| PARP11 | GO:0003950 | Actividad NAD + ADP- ribosiltransferasa |
Función molecular |
| IFI44L* | GO:0000107 | Proteína que contiene el dominio TLDc |
Procesos biológicos |
| PTGFR | GO:0004958 | Actividad del receptor de prostaglandina F |
Función molecular |
| NRXN3 | |||
| ATXN1 | GO:0045892 | Regulación negativa de la transcripción, plantilla de ADN |
Procesos biológicos |
| MANBA | GO:0005975 | Proceso metabólico de carbohidratos |
Procesos biológicos |
| NFKB1 | GO:0006357 | Regulación de la
transcripción por la ARN polimerasa II |
Procesos
biológicos |
A continuación, se describen 16 genes candidatos identificados en regiones con SS en CPM. 1) ENSSSCG00000007733, se considera un gen nuevo involucrado en las hernias umbilicales27. 2) VIT Vitrin, relacionado con hernias escrotales y umbilicales28,29. 3) RAC2 Sustrato 2 de toxina botulínica C3, es una GTPasa que se expresa en células hematopoyéticas con funciones en los leucocitos y neutrófilos30, participa en la regulación de distintas rutas de regulación del ciclo celular, apoptosis y respuesta inmune31. 4) SSTR3, es uno de los cinco genes que codifican para los receptores de la somatotropina y se expresa en la pituitaria32. 5) C1QTNF6, proteína 6 relacionada con el factor de necrosis tumoral, controla la deposición de grasa subcutánea e intramuscular por medio de las vías de señalización MAPK, p53, TNF y adipocinas33. 6) TMPRSS6, serina proteasa transmembrana de tipo dos, matriptasa - 2, regulador proteolítico de la homeostasis del hierro34. 7) CRACR2A, regulador 2A del canal activado de liberación de calcio; GTPasa, actúa en la transmisión de señales entre la estimulación del receptor de las células T y la activación de las rutas Ca2+-NFAT y JNK-AP135. 8) PARP11, miembro 11 de la familia de las polimerasas ADP - ribosiltransferasa poli (ADP-ribosa), importante regulador de la eficiencia antiviral del IFN-136. 9) ENSSSCG00000029668 (IL2RB), receptor de interleucina-2, las acciones pleiotrópicas son esenciales para la regulación de la respuesta inmune y del mantenimiento de la tolerancia inmunitaria37. 10) PTGFR, receptor de prostaglandina F2α38. 11) IFI44L, es un gen inducible especifico de interferón, puede producir un agotamiento de GTP celular, suprimiendo la señalización de la quinasa regulada por señales extracelulares deteniendo el ciclo celular39. 12) NRXN3 neurexina 3, gen candidato de susceptibilidad a trastornos del neurodesarrollo, las mutaciones modifican la estabilidad y función sináptica; se sintetiza en todas las neuronas excitadoras e inhibidoras40,41. 13) ATXN1 ataxina-1, la expresión anormal causa la enfermedad neurodegenerativa ataxia espinocerebelosa tipo 142. 14) CCSER1, es un gen de proteína reguladora, asociado a rasgos económicos como crecimiento, desarrollo, eficiencia alimenticia y calidad de la leche43-46. 15) MANBA beta - manosidasa, desempeña funciones importantes en la regulación del sistema inmunológico47,48. 16) NFKB1, factor nuclear del potenciador del gen del polipéptido ligero kappa en células B1, reguladoras de la inmunidad, proliferación celular, respuesta al estrés y apoptosis49.
En la definición de una raza las marcas genéticas buscan regiones centrales positivas, inmersas a la selección artificial y natural en su entorno50,51. Las SS se derivan de procesos de selección que han aportado a la adaptación en distintos entornos y sistemas productivos52. A diferencia de las razas especializadas, en el CPM la selección no ha sido dirigida a parámetros de interés económico, sino a la conservación, adaptación y evolución misma. La selección en programas de conservación se desarrolla con criterios asociados a estándares de la raza, dirigida a la consolidación de rasgos específicos y estabilización del fenotipo, con cambios menos pronunciados en la frecuencia de los alelos relacionados5. Las SS en razas autóctonas se asocian con la domesticación y el establecimiento de estándares raciales, no con parámetros de interés económico dados por selección artificial53. Con relación a la longitud del haplotipo, se relaciona con selección natural (regiones cortas) o selección artificial; no obstante, no existe una línea especifica que divida el tipo de selección, pueden estar influenciadas entre sí y existir regiones superpuestas en el genoma50. Con respecto a razas autóctonas, Muñoz et al53 identificaron SS en los cromosomas en razas europeas, con GC asociados a reproducción, comportamiento locomotor, metabolismo de lípidos, crecimiento y desarrollo; también señalan que el cromosoma 7 está asociado con procesos de la domesticación y con rasgos relacionados con el comportamiento animal. El cromosoma 8 ha sido referenciado con características de crecimiento, altura y conformación52,54, así como GC con la respuesta a enfermedades inmunes55.
El cromosoma 7 (Figura 2) presentó un haplotipo corto y degradado, el cual se puede relacionar a procesos de adaptación; el cromosoma 8 mostró un haplotipo largo y conservado, asociado a una selección reciente dirigida a parámetros morfológicos. En cerdos Laiwu, raza autóctona reconocida por su alto contenido de grasa intramuscular12,56, se han reportado SS asociando GC a características reproductivas, de desarrollo, crecimiento, gordura, ganancia de peso, consumo de alimento, respuesta inmune, deposición de grasa y tamaño de camada. Guo et al50 identificaron GC asociados a la resistencia de enfermedades, rasgos reproductivos y calidad de la carne, como resultado de la detección de SS en el genoma de una población de cerdos indígenas de Anqing en China. Chen et al57 reportaron QTL relacionados con el grosor de la grasa dorsal, color de la carne, valor del pH, contenido de ácidos grasos, células inmunes, inmunidad parasitaria e inmunidad bacteriana. Wang et al58 identificaron SS en una población de cerdos Tunchang, con GC relacionados con adaptabilidad, resistencia a enfermedades y rasgos de metabolismo de lípidos; con estas características, los investigadores discuten que la raza Tunchang está más relacionada con cerdos autóctonos que con razas especializadas. Quin et al59 evaluaron regiones en el genoma que sufrieron selección asociadas a GC que regulan la inmunidad y la deposición de grasa en cerdos autóctonos de Shandong China. Gurgul et al5 estudiaron razas de cerdos en programas de conservación en Polonia, donde detectaron varias SS con GC asociados a vías metabólicas, respuesta del sistema inmunológico y desarrollo embrionario. En razas ibéricas y Casertanas, los QTL y los escaneos en el genoma han identificado regiones y mutaciones relacionadas con aspectos morfológicos, parámetros productivos, salud, características de carne y canal52,60. Para cerdos autóctonos de Huainan y su cruza con la raza Duroc se han identificado SS, donde se asociaron QTL para contenido de grasa intramuscular en la raza autóctona y para la cruza se asociaron para el contenido de triglicéridos, cerdos momificados, hemoglobina y profundidad del músculo del lomo; la detección de SS es una herramienta viable para la evaluación del rendimiento productivo en el cruzamiento de cerdos autóctonos con razas especializadas61, por lo que la información genómica juega un papel importante en la mejora de programas de conservación62.
Se concluye que el CPM sufrió selección, y se identificaron GC y QTL asociados a la respuesta del sistema inmune, adaptación, comportamiento, obesidad, implantación y desarrollo embrionario, calidad de la carne por la grasa intramuscular, crecimiento, desarrollo y eficiencia alimenticia, así como características morfológicas que lo pueden distinguir de otras razas; también se observó que se seleccionaron indirectamente genes candidatos indeseables con relación a hernias umbilical y escrotal.

