











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Information theory, as originally developed by Claude Shannon [1] to study communication systems is referred as to classical information theory (CIT) and it has found significant applications in chemistry due to its ability to quantify and analyze information in chemical processes and molecular systems. It provides tools for quantifying uncertainty and information content in chemical systems, aiding in the understanding of molecular interactions and reactions at a fundamental level [2]. By applying concepts such as entropy and mutual information, chemists gain insights into the structure and dynamics of molecules, which is crucial for predicting molecular behavior, interactions, and designing new materials and drugs [3]. Information theory techniques are also used to compress large datasets from chemical experiments and simulations, making data storage and transmission more efficient while reducing noise in experimental data for more accurate results [4]. It bridges thermodynamics and statistical mechanics by providing a formalism to describe the distribution of states in a system, leading to a deeper understanding of thermodynamic properties and phase transitions [5]. In computational chemistry, information theory helps optimize algorithms for molecular simulations and quantum calculations, playing a significant role in machine learning applications and enhancing the analysis and prediction of chemical phenomena [6]. Additionally, chemical informatics, which involves the storage, retrieval, and analysis of chemical information, heavily relies on information theory to improve the organization and interpretation of chemical data, facilitating advancements in research and industry [7]. Thus, information theory enriches chemistry by providing a rigorous framework for analyzing and interpreting chemical information, leading to deeper insights and more efficient methodologies in research and application.
In contrast to CIT, quantum information theory (QIT) [8] is an interdisciplinary field spanning theoretical physics and computer science that investigates how the fundamental principles of quantum mechanics can be utilized for information processing, transmission, and storage in ways that radically diverge from classical information theory. QIT in essence, examines the potential of quantum mechanical concepts and properties, such as superposition, entanglement, and uncertainty, to develop new ways to encode, manipulate, and communicate information that are unattainable for CIT within the classical realm governed by the laws of classical physics and traditional information theory.
Quantum information chemistry (QIChem) is an emerging field that combines quantum information science with chemistry to study and manipulate chemical systems at the quantum level [9]. This interdisciplinary approach promises to revolutionize our understanding of chemical processes and drive advancements in various scientific and industrial applications. At its core is the use of quantum computers [11], which harness quantum mechanics to solve complex chemical problems intractable for classical computers. These quantum computers offer unprecedented accuracy and efficiency in simulating molecular structures, reaction mechanisms, and energy states - capabilities crucial for predicting molecular behavior and designing new compounds.
Quantum simulations enable detailed and accurate modeling of quantum-level chemical processes, essential for drug discovery, materials science, and catalysis [12]. Quantum algorithms like Quantum Phase Estimation (QPE) and the Variational Quantum Eigensolver (VQE) [13] are designed to efficiently determine electronic structures of molecules by utilizing quantum superposition and entanglement. Moreover, quantum control techniques [14] manipulate quantum systems to achieve desired chemical outcomes, influencing reactions and bond formations for novel synthetic pathways and improved efficiencies. Applying quantum information concepts like entanglement and superposition provides new insights by elucidating the effects of quantum coherence on chemical dynamics [15].
The applications of QIChem are diverse, spanning drug discovery via simulating molecular interactions [16], materials design by predicting molecular structures and behaviors [17], improved catalysis through deeper understanding of catalyst-reactant interactions [18], energy storage technologies benefiting from insights into quantum material properties involved in energy transfer [19], and environmental solutions via simulations for pollution control and resource management [20].
However, challenges persist, including current quantum hardware limitations like high error rates, limited qubit counts, and the need for advanced algorithms to model large, complex molecules [21]. Overcoming these obstacles necessitates collaboration between quantum physicists, chemists, and computer scientists. This rapidly evolving field aims to improve quantum algorithms, develop more stable and scalable hardware, and discover new applications in chemistry and materials science [22]. QIChem has the potential to fundamentally transform our understanding and manipulation of chemical processes by leveraging quantum computing and mechanics for complex chemical problems, promising significant advancements in drug discovery, materials science, catalysis, energy storage, and environmental chemistry [23]. Despite ongoing challenges, research and technological developments could revolutionize the approach to chemical research and industrial applications.
Overview
This review explores the local aspects of the classical information (CIT) such as spreading, ordering, disequilibrium and complexity but also the non-local phenomena associated with entanglement (non-locality) through the quantum information-theoretic concepts (QIT) associated to the superposition of the states. These phenomena are analyzed on a variety of chemical systems, from atoms, ions, molecules to amino acids unveiling fundamental aspects of the chemical systems and the processes they exert, towards developing the emerging field of Quantum Information Chemistry (QIChem).
Here some advances and novel interpretations:
Shannon entropy analysis has predicted reaction mechanisms for elementary reactions of SN2 and hydrogenic abstraction type. We identified chemically important regions such as reactant/product (R/P) complexes, the transition state (TS), and others revealed only through IT measures, like the bond-cleavage energy region (BCER), bond-breaking/forming (BB/F) region, and spin-coupling (SC) process.
Quantifying complexity in physical, chemical, and biological systems is achievable through information-theoretic measures like Shannon entropy (S), Fisher information (I) along with disequilibrium (D), altogether help describing the complexity and information behavior of systems.
Our research has also been focused to explore elementary chemical reactions using information theory in 3D space, focusing on functionals like disequilibrium, Shannon entropy, Fisher information, and complexity measures such as Fisher−Shannon (FS) and López−Mancini−Calbet (LMC). The study uses the hydrogenic identity abstraction reaction to examine reactivity patterns through these functionals in both position (r) and momentum (p) spaces. The analysis reveals significant reactivity patterns around the intrinsic reaction coordinate (IRC) path, offering new insights into the reaction mechanism.
Information Theory analysis in 3D space of the dissociation process of the triatomic transition-state complex formed at the transition state of the hydrogenic identity abstraction reaction highlights interesting features of bond-breaking (B-B). The study shows that chemical reactions occur in low-complexity regions where various phenomena like B-B/F, BCER, SC, and TS converge.
A pioneering information-theoretical analysis of 18 amino acids from bacteriorhodopsin (1C3W) was conducted. The Shannon entropy, Fisher information, and disequilibrium were used to characterize amino acids by their delocalizability, order, and uniformity, forming a scheme that classifies them into four major families: aliphatic, aromatic, electro-attractive, and tiny.
We propose a 3D Information-Theoretic Chemical Space using Shannon entropy, Fisher information, and disequilibrium measures to unveil unique aspects of many-electron systems, from simple molecules to complex systems like amino acids and pharmacological molecules. This space is based on the fundamental information-theoretic notions of delocalization, order, uniformity, and complexity.
We explore the partitioning of molecules into constituent parts using Atoms-In-Molecules (AIM) schemes and their information-theoretic justifications. We validated popular AIM schemes like Hirshfeld, Bader's topological dissection, and the quantum approach within this framework.
We discuss the quantum origin of correlation energy, relating it to entanglement as measured through von Neumann and linear entropies. For helium-like systems, a direct correlation between entanglement and correlation energy was observed. We provided numerical evidence of a linear relationship between correlation energy and quantum entanglement for the helium isoelectronic series.
The study also delves into quantum entanglement aspects of the dissociation processes in homo- and heteronuclear diatomic molecules, using high-quality ab initio calculations. The behavior of electronic entanglement along the reaction coordinate aligns with significant physical changes in molecular density.
Finally, we investigate quantum entanglement, a profound manifestation of non-classical correlations between subsystems, suggesting that chemical systems inherently possess some level of entanglement, underscoring the importance of exploring its potential connection to chemical stability and reactivity.
The present review discusses the phenomenological description of several systems of higher complexity, from atoms and molecules of biological interest to dendrimers of nanotechnological nature. Furthermore, chemical transformations associated with reactivity theories are studied from the theoretical perspective of the “local” and “non-local” aspects of the pure state electron densities associated with those processes and the mixed states linked to the wavefunction superposition of the density matrix, respectively. This approach permits to employ two different information-theoretic schemes: (i) the classical description (locality) of chemical phenomena through CIT and also, (ii) the quantum effects associated to entanglement (non-locality) by use of QIT.
Henceforth, the analyses are separated into local and non-local phenomena covering several systems of higher complexity and processes associated with different type of chemical transformations:
Local Phenomena
Chemical Processes
Phenomenological Description of Two Center Reactions
Radical Abstraction Reaction (SN1)
Nucleophilic Substitution Reaction (SN2)
3D Complexity analysis of the Radical Abstraction Reaction
Molecules
Complexity and Information Planes of Selected Molecules
Predominant Information-Theoretic Quality scheme (PIQS) of amino acids
3D Information-Theoretic Chemical Space from Atoms to Molecules
The Separability Problem: Atoms in Molecules in Fuzzy and Disjoint Domains
Non-Local Phenomena
Quantum Entanglement
Correlation energy as a measure of non-locality: helium-like systems
Dissociation Process of Diatomic Molecules
Chemical Reactivity
Concluding Remarks
Chemical Processes
Theoretic-information measures of the Shannon type have been employed to describe the simplest hydrogen abstraction SN1 and the identity SN2 exchange chemical reactions [24]. These measures effectively detect the transition state and reveal bond breaking/forming regions. A numerical verification supports the argument that information entropy profiles possess more chemically meaningful structure than total energy profiles for these reactions. The results align with Zewail and Polanyi's concept of a continuum of transient states for the transition state, supported by reaction force analyses. Additionally, the information-theoretic description using Shannon entropic measures in position and momentum spaces allows a phenomenological description of these reactions' chemical behavior, revealing synchronous and asynchronous mechanistic behavior [25].
Informational-theoretic measures have also been applied to describe the course of a three-center insertion reaction [26], identifying transition states and stationary points that reveal bond breaking/forming regions not evident in the energy profile. These results further support the continuum of transient states concept. Fisher information measures can scrutinize the local behavior of chemically significant distributions in conjugated spaces, detecting transition states, stationary points, and bond breaking/forming regions for elementary reactions like the simplest hydrogen abstraction and identity SN2 exchange reactions [27].
The origin of the internal rotation barrier between eclipsed and staggered conformers of ethane has been systematically investigated from an information-theoretical perspective using the Fisher information measure in conjugated spaces [28]. Adiabatic (optimal structure) and vertical (fixed geometry) computational approaches were considered, following the conformeric path by changing the dihedral angle. The results showed that in the adiabatic case, the eclipsed conformer has larger steric repulsion than the staggered conformer, while in the vertical cases, the staggered conformer retains larger steric repulsion. These findings verify the feasibility of defining and computing the steric effect at the post-Hartree-Fock level of theory according to Liu's scheme.
Statistical complexity is of growing interest in physical sciences. A recent investigation examined the complexity of the hydrogenic abstraction reaction [29] using information functionals such as disequilibrium ( D ), exponential entropy ( L ), Fisher information ( I ), power entropy ( J ), and joint information-theoretic measures, including the I-D, D-L, and I-J planes and the Fisher-Shannon (FS) and LMC shape complexities. The analysis identified all chemically significant regions from the information functionals and most information-theoretical planes, including reactant/product regions (R/P), transition state (TS), bond cleavage energy region (BCER), and bond breaking/forming regions (B-B/F), which are not present in the energy profile. The complexities analysis showed that the energy profile of the abstraction reaction bears the same information-theoretical features of the LMC and FS measures in position and joint spaces. The study explained why most chemical features of interest, such as BCER and B-B/F, are absent in the energy profile and only revealed when specific information-theoretical aspects of localizability ( L or J ), uniformity ( D ), and disorder (I ) are considered.
Some other topics of interest have been incorporated in this part of the Review article: (i) concurrent phenomena occurring at the vicinity of transient state, (ii) and the Hypersurfaces of the information-theoretic functionals and complexity measures in 3D space for the radical abstraction reaction.
Phenomenological description of two center reactions
Predicting molecular structure and energetics during dissociations or chemical reactions is a major focus in theoretical and computational chemistry, spanning multiple research areas and driving the development of numerous theories and models extensively discussed in the literature [30]. A key aspect involves characterizing chemical processes in terms of physical phenomena like charge redistribution, bond breaking/forming, and reaction pathways.
Potential energy surface (PES) calculations at various levels of theory have been widely employed to understand the stereochemical course of chemical reactions [31]. These studies extract information about stationary points on the energy surface. According to the Born-Oppenheimer approximation, minima on the N-dimensional PES correspond to equilibrium molecular structures, while saddle points correspond to transition states and reaction rates. Since the inception of transition-state (TS) theory [32], significant efforts have focused on developing models to characterize the TS, crucial for understanding chemical reactivity.
Computational quantum chemistry addresses these challenges by defining "critical points" on a potential energy hypersurface, identifying equilibrium complexes or transition states. This approach utilizes first and second derivatives of energy (gradient and Hessian) relative to nuclear positions. Multiple minima on a contiguous energy surface allow for the construction of reaction paths, with the TS defined as the lowest maximum along these paths and a first-order saddle point. The eigenvector of the single negative eigenvalue at this critical point is the transition vector, guiding the steepest-descent path from the saddle point to the minima (reactants or products).
The unique reaction path can be defined using an intrinsic reaction coordinate (IRC), independent of the coordinate system, by employing classical mechanics in mass-weighted Cartesian coordinates [34]. The IRC is the path traced by a classical particle moving with infinitesimal velocity from a saddle point to the minima, aligning with the steepest-descent path in mass-weighted coordinates. Several computational techniques have been developed to calculate energy gradients and Hessians, allowing the following of such reaction paths [35].
Despite the utility of critical points on the energy surface for analyzing reaction paths, their chemical or physical significance remains uncertain [36]. The pursuit to understand the transition state (TS) structure represents a challenge in physical organic chemistry, although chemical concepts like reaction rate and barrier have been extensively studied. Efforts to achieve this have produced chemically useful TS descriptions, such as the Hammond-Leffler postulate [37]. Hammond proposed that points on a reaction profile with similar energies will also have similar structures, allowing TS structure predictions in highly exothermic and endothermic reactions. Leffler generalized this idea, considering the TS as a hybrid of reactants and products with an intermediate character. The Hammond-Leffler postulate remains a practical tool, stating that TS properties are intermediate between reactants and products, related to its position along the reaction coordinate.
With the advent of femtosecond time-resolved methods, these theories have gained renewed relevance. Since Zewail and co-workers' seminal studies [38], femtochemistry techniques have been applied to chemical reactions of varying complexity, providing new insights into fundamental chemical processes. As Zewail stated, femtochemistry allows observing "the very act of breaking or making a chemical bond" by "freezing" the molecular dynamics. Although most femtochemistry studies deal with excited states, ground-state processes have been studied as well. One promising technique, anion photodetachment spectra [39], has enabled direct observation of transition states. To explain the experimental results of femto-techniques, it will be necessary to complement existing chemical reactivity theories with electronic density descriptors of events occurring in the transition-state region, where bonds are formed or broken.
Numerous studies have employed various descriptors to investigate transition state (TS) structure or follow the chemical reaction path. Shi and Boyd systematically analyzed model SN2 reactions to study TS charge distribution in connection with the Hammond-Leffler postulate [40]. Bader et al. developed a reactivity theory based on charge density properties by employing the Laplacian, aligning charge concentrations with depletion regions by mixing in the lowest excited state to produce a transition density [41]. Balakrishnan et al. showed information-theoretic entropies in phase space peaked during a bimolecular exchange reaction [42]. Ho et al. found information measures revealed geometrical density changes not present in the energy profile for SN2 reactions, though not identifying TSs [43]. Knoerr et al. correlated charge density features with energy-based charge transfer [44], stability, and localization measures for an SN2 reaction, attempting a density-based reactivity theory [45]. Tachibana visualized bond formation using kinetic energy density to identify reactant, TS, and product shapes along the IRC [46], realizing Coulson's conjecture [47]. Reaction forces along the reaction coordinate have characterized structural/electronic changes [48]. The Kullback-Leibler deficiency has been evaluated along internal coordinates and IRCs for SN2 reactions [49].
In spite of the interest in applying information theory (IT) measures to electronic structure [50], their effectiveness as descriptors for characterizing IRC stationary points (TS, equilibrium geometries) and bond breaking/forming regions remained unclear. Indeed, proper analysis of IT descriptors (Ref [24]) unveil the phenomenological behavior of elementary reactions by following IRCs, analyzing density behavior in position/momentum spaces near the TS and bond breaking/forming regions not visible in the energy profile using Shannon entropies. Charge density descriptors like the molecular electrostatic potential (MEP), density functional theory hardness/softness will link density changes to information quantities during reactions.
The chemical probes under study are the simplest hydrogen abstraction reaction
The central quantities under study are the Shannon entropies in position and momentum spaces [1]:
where ρ(r) and γ(p) denote the molecular electron density distributions in the
position and momentum spaces each normalized to unity. In the
independent-particle approximation, the total density distribution in a molecule
is a sum of contribution from the electrons in each of the occupied orbitals.
This is the case in both r-space and p-space,
position and momentum respectively. In momentum space, the total electron
density,
The Shannon entropy in position space Sr behaves like a measure of delocalization or lack of structure of the electronic density in the position space and hence Sr is maximal when knowledge of ρ(r) is minimal and becomes delocalized. The Shannon entropy in momentum space Sp is largest for systems with electrons of higher speed (delocalized γ(p)) and is smaller for relaxed systems where kinetic energy is low. Entropy in momentum space Sp is closely related to Sr by the uncertainty relation of Bialynicki-Birula and Mycielski [53], which shows that the entropy sum ST=Sr+Sp, is a balanced measure and cannot decrease arbitrarily. For one-electron atomic systems it may be interpreted as that localization of the electron’s position results in an increase of the kinetic energy and a delocalization of the momentum density, and conversely.
Regarding the behavior of the Shannon entropy discussed, a simple explanation is as follows: Since there is no variational principle for quantum mechanical properties besides the energy, directly relating the Shannon entropy to the transition state seems impractical. However, the plausible argument is that the transition state should correspond to a highly localized momentum distribution and delocalized position distribution per the uncertainty principle. This localized momentum would represent minimum kinetic energy as per classical Mechanics. The main points are: a) The highly localized momentum/delocalized position at the transition state corresponds to minimum kinetic energy, as understood by classical Mechanics, and b) this highly localized momentum/delocalized position corresponds to the minimum kinetic energy in the classical understanding.
To summarize, the highly localized momentum in the classical understanding represents the minimum kinetic energy, which matches the arguments of minimum kinetic energy in the classical understanding. The key distinction is that the highly localized momentum represents the minimum kinetic energy in the classical interpretation/understanding, which is consistent with the high degree of momentum localization discussed. The difference is the classical interpretation/understanding of the localized momentum representing minimum kinetic energy.
The MEP represents the molecular potential energy of a proton at a particular location near a molecule [54], say at nucleus A. Then the electrostatic potential, VA, is defined as
where ρ(r) is the molecular electron density and ZA is the nuclear charge of atom A, located at RA. Generally speaking, negative electrostatic potential corresponds to an attraction of the proton by the concentrated electron density in the molecules from lone pairs, pi-bonds, etc… (coloured in shades of red in standard contour diagrams). Positive electrostatic potential corresponds to repulsion of the proton by the atomic nuclei in regions where low electron density exists and the nuclear charge is incompletely shielded (coloured in shades of blue in standard contour diagrams).
We have also evaluated some reactivity parameters that may be useful to analyze the chemical reactivity of the processes. Parr and Pearson, proposed a quantitative definition of hardness (() within conceptual DFT [55]:
where
where I and A, are the ionization potential (IP) and electron affinity (EA), respectively. Applying Koopmans’ theorem [56], Eq. (4) can be written as:
where ( denotes the frontier molecular orbital energies. In general terms, hardness and softness are good descriptors of chemical reactivity, the former measures the global stability of the molecule (larger values of η means less reactive molecules), whereas the S index quantifies the polarizability of the molecule [57], thus soft molecules are more polarizable and possess predisposition to acquire additional electronic charge [58]. The chemical hardness “η” is a central quantity for use in the study of reactivity and stability, through the hard and soft acids and bases principle [59]. However, in many cases, the experimental electron affinity is negative rather than positive, an such systems pose a fundamental problem; the anion is unstable with respect to electron loss and cannot be described by a standard DFT ground-state total energy calculation. To circumvent this limitation, Tozer and De Proft have introduced an approximate method to compute this quantity, requiring only the calculation of the neutral and cationic systems which does not explicitly involve the electron affinity [60]:
where I is obtained from total electronic energy calculations on the N-1 and N
electron systems at the neutral geometry
The electronic structure calculations were conducted using the Gaussian 03 software suite [62]. Reported transition state (TS) geometries from previous studies were utilized for the abstraction [62] and SN2 exchange reactions [63]. Internal reaction coordinate (IRC) calculations [64] along the forward and reverse reaction paths were performed at the MP2 level of theory (UMP2 for the abstraction reaction), with a minimum of 35 points along each IRC path direction. A high level of theory and well-balanced basis set, incorporating diffuse and polarized orbitals, were selected for computing all properties of the structures along the IRC path. The chemical hardness and softness parameters were evaluated using Eqs. (6) and (7) with the standard hybrid B3LYP functional (UB3LYP for the abstraction reaction) [60].
Molecular vibrational frequencies, derived from the eigenvalues of the Hessian matrix (whose elements are associated with force constants) at the stationary point nuclear coordinates, were calculated. Notably, the values corresponding to the normal mode at the TS (exhibiting one imaginary frequency or negative force constant) were determined analytically at all points along the IRC path using the MP2 level of theory (UMP2 for the abstraction reaction) [64]. The molecular information entropies in position and momentum spaces along the IRC path were obtained using in-house software incorporating 3D numerical integration routines [65], along with the DGRID program suite [66]. The bond breaking/forming regions, as well as electrophilic/nucleophilic atomic regions, were evaluated through the molecular electrostatic potential (MEP) using the MOLDEN software [67]. Unless stated otherwise, atomic units were employed throughout the study.
Radical abstraction reaction
The reaction
For this reaction, calculations were performed at two different levels of theory. The internal reaction coordinate (IRC) was obtained at the UMP2/6-311G level, while all properties along the IRC path were computed at the QCISD(T)/6-311++G** level. As a result of the IRC calculation, 72 points were evenly distributed between the forward and reverse directions of the reaction. A relative tolerance of 10-5 was set for the numerical integrations [66].
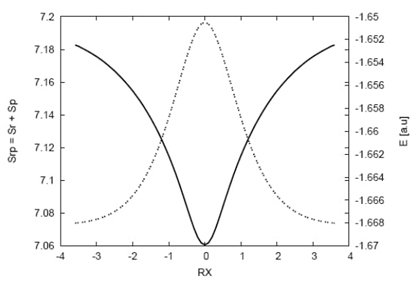
Fig. 1 Total energy values (dashed line) in a.u. and the entropy sum (solid line) for the IRC path of the hydrogenic abstraction reaction.
The energy profile and entropy sum along the intrinsic reaction coordinate (IRC) path for the abstraction reaction are shown in Figure 1. The energy profile exhibits symmetric behavior, while the entropy sum (TS) shows the opposite trend. The saddle point structure has a localized density in the combined position and momentum space (phase space), corresponding to a more delocalized position density with the lowest kinetic energy among the nearby structures along the IRC path. This suggests that the saddle point can be characterized by information theory (IT) in the entropy hyper-surface.
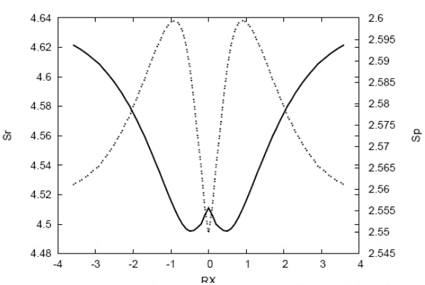
Fig. 2 Shannon entropies in position (solid line) and momentum (dashed line) spaces for the IRC path of the hydrogenic abstraction reaction.
Fig. 2 presents the Shannon entropies in position and momentum spaces along the IRC path. The position entropy has a local maximum at the transition state (TS) and two minima in its vicinity, while the momentum entropy shows the opposite behavior with a minimum at the TS and two maxima nearby. As the intermediate radical approaches the molecule, the position entropy decreases towards the TS region, indicating that the densities of the chemical structures are more localized in this region, where important chemical changes occur. In contrast, the momentum entropy increases near the TS, which is linked to a more localized momentum density with the lowest kinetic energy (corresponding to the maximum on the potential energy surface). At the reactive complex regions (towards reactants and products), the momentum entropy and kinetic energy values are higher compared to the TS, reproducing the typical potential energy surface shown in Fig. 1.
In summary, the entropy and energy profiles provide valuable insights into the chemical changes occurring along the IRC path, with the TS region characterized by localized densities in position space and delocalized densities in momentum space, reflecting the lowest kinetic energy and the highest potential energy at the saddle point.
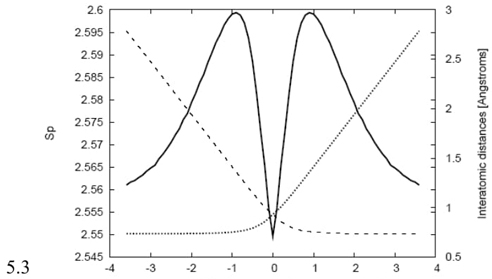
Fig. 3 Shannon entropy in momentum space (solid line) and the bond distances R(H0-Hin) (dashed line for the entering hydrogen) and R(H0-Hout) (dotted line for the leaving hydrogen) in Angstroms for the IRC path of the hydrogenic abstraction reaction.
Fig. 3 illustrates the bond distances between the entering/leaving hydrogen radicals and the central hydrogen atom along the reaction path. The plot clearly indicates that in the vicinity of the transition state (TS), a bond breaking and forming process is taking place. On the right side of the TS, the bond distance Rin is elongating, signifying the breaking of the bond. Simultaneously, on the left side of the TS, the bond distance Rout is stretching, indicating the formation of a new bond. Interestingly, the chemical process does not occur in a concerted manner. Instead, the reaction follows a two-step mechanism. First, homolytic bond cleavage takes place, and then the molecule stabilizes by forming the TS structure. This stepwise process is evident in Fig. 3. As the incoming radical approaches the molecule, the bond breaks at the same location where the position entropy reaches its minimum value, and the momentum entropy attains its maximum. This observation aligns with the previous discussion regarding the localized density in position space and delocalized density in momentum space at the TS region. After the bond cleavage and TS formation, the new molecule is formed, completing the reaction process. The analysis of bond distances and the entropy profiles supports the two-step mechanism characterizing this abstraction reaction.
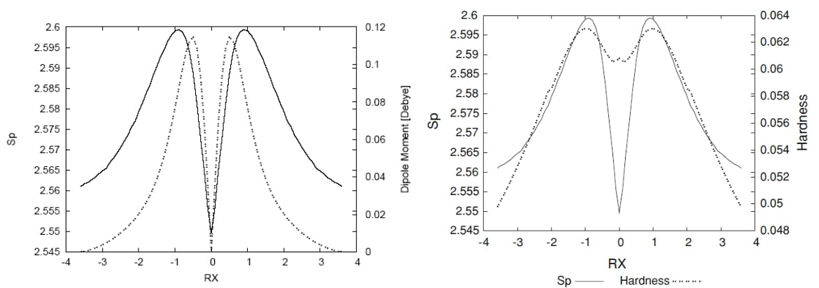
Fig. 4 Shannon entropy in momentum space (solid line), the dipole moment values in Debyes (dashed line), and the hardness values (dashed line) for the IRC path ofthe hydrogenic sbstraction reaction.
In Fig. 4, the non-polar bond pattern characteristics of homolytic bond-breaking reactions are studied by examining the dipole moment and momentum entropy along the intrinsic reaction coordinate (IRC) path [24]. The dipole moment is zero at the transition state (TS) and when approaching reactants/products, reflecting the non-polar nature of the molecule in these regions. However, the molecular densities undergo significant distortion near the TS, where position entropies are minimal, indicating a rigid molecular geometry with localized position density at the bond breaking/forming regions. The momentum entropy maxima reveal that the energy reservoirs for bond cleavage, referred to as bond cleavage energy reservoirs (BCER), occur earlier or later along the IRC path, depending on the reaction direction. The interplay between dipole moment, position entropy, and momentum entropy provides insights into the complex nature of the reaction and the critical role of the TS and BCER regions in the overall mechanism. Also, in Fig. 4 we have depicted the hardness values along with the momentum space entropy for comparison purposes. From a DFT conceptual point of view we may interpret Fig. 4 as that chemical structures at the maximal hardness (minimal softness) values possess low polarizability and hence are less propense to acquire additional charge (less reactive). According to considerations discussed above, these structures are found at the BCER regions, i.e., they are maximally distorted, with highly localized position densities (minimal position entropies, and maximal dipole moment values, (see Fig. 4). In contrast, hardness values are maximal at the reactant/product complex regions which correspond with delocalized position densities with null dipole moments, hence they are more prone to react (more reactive). At the TS, a local minimum for the hardness may be observed, then it is locally more reactive and leading to acquire charge since its dipole moment is null. Accordingly, the TS structure is more relaxed and with a more delocalized density.
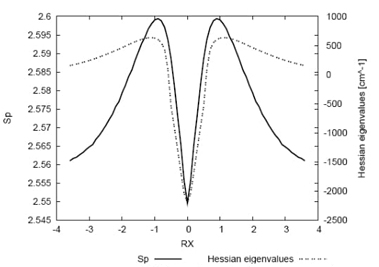
Fig. 5 Shannon entropy in momentum space (solid line) and the
eigenvalues of the Hessian (dashed line) for the IRC path of
In Fig. 5, the eigenvalues of the Hessian matrix for the normal mode associated with the transition state (TS) are plotted along the reaction path, together with the momentum entropy values for comparison. These Hessian values signify the transition vector "frequencies," exhibiting maxima in the vicinity of the TS and a minimum value at the TS itself. Several noteworthy aspects emerge from this analysis. Firstly, the TS indeed represents a saddle point, with the Hessian maxima corresponding to high kinetic energy values (largest "frequencies" for the energy cleavage reservoirs), aligning with the peak values in the momentum entropy profile. Conversely, the Hessian is minimal at the TS, where the kinetic energy is the lowest (minimal molecular frequency), coinciding with a minimal momentum-entropy value. Additionally, the transition state of a reaction is typically characterized by the presence of a negative force constant for one normal vibrational mode, associated with an imaginary frequency. The pioneering work of Zewail and Polanyi in transition state spectroscopy has introduced the concept of a reaction possessing a continuum of transient states, referred to as a transition region, rather than a single transition state [38,68]. Remarkably, the findings of the current study provide evidence for the existence of such a region between the bond cleavage energy reservoirs (BCER) situated before and after the TS. This is in agreement with reaction force, F(R), studies [48] where the reaction force constant, κ(R), also reflects this continuum, showing it to be bounded by the minimum and the maximum of F(R), at which κ(R) = 0.
The reactivity characteristics of the reaction have also been explored using density-based descriptors, such as hardness and softness, within the framework of conceptual density functional theory (DFT) [24]. According to this approach, chemical structures exhibiting maximum hardness (and, consequently, minimum softness) have low polarizability, making them less likely to accept additional charge and, thus, less reactive. Interestingly, these structures are located in the bond cleavage energy reservoir (BCER) regions, where they are maximally distorted and possess highly localized position densities, see Fig. 6 in Ref [24].
Hydrogenic identity SN2 exchange reaction
In the study of elementary chemical reactions, analyzing a typical nucleophilic substitution (SN2) reaction is of particular interest due to its single-step process, unlike the two-step SN1 reaction. The anionic SN2 mechanism, depicted as Y− + RX → RY + X−, is second-order kinetics, being first order in both the nucleophile (Y−) and the substrate (RX, where X− is the leaving group). For identity SN2 reactions, X=Y. The observed second-order kinetics result from the Walden inversion transition state, where the nucleophile displaces the leaving group from the backside in a single, concerted reaction step. Evidence indicates that this reaction mechanism is characterized by synchronous and concerted behavior [25].
The
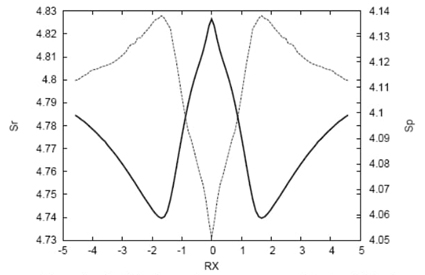
Fig. 6 Shannon entropies in position (solid line) and momentum (dashed line) spaces for the IRC path of the SN2 reaction at the QCISD(T)/6-311++G** level.
A comparison between the entropy sum (Fig. 8 in Ref [24]) and the energy reveals contrasting behaviors, with the entropy sum exhibiting much more structure near the transition state (TS) region compared to the energy profile [24]. The nature of the richer structure observed for the entropy sum, in contrast to the energy, is elucidated through the position and momentum entropies (illustrated in Fig. 6). These show a TS structure characterized by a delocalized position density and a localized momentum density, indicating a structurally relaxed configuration with low kinetic energy. In contrast, towards the reactive complex and reactants/products, the position densities are more localized with less localized momentum densities, signifying structurally distorted chemical configurations with higher kinetic energy compared to the TS. Around |RX| ≈ 1.7, critical points for both entropies are observed, with minima/maxima for the position/momentum entropies, respectively. Thus, ionic complexes in these regions are characterized by highly localized position densities and highly delocalized momentum densities, along with high kinetic energies, suggesting that these regions correspond to bond critical energy regions (BCER) where bond breaking may initiate. Two additional features are noteworthy: both entropies exhibit inflection points at |RX| ≈ 1.0 and maxima at |RX| ≈ 0.5. These are regions where the entropy sum displays more defined structure, with changes in curvature and maxima, respectively [24]. Further exploration of these observations in connection with other properties will be conducted later.
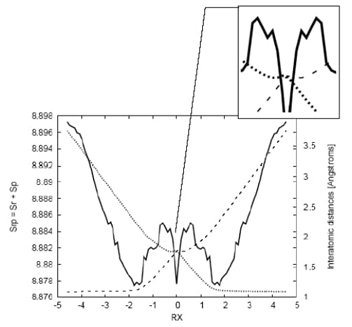
Fig. 7 Shannon entropy in momentum space (solid line) and the bond distance Ra (dotted line), corresponding to the Ha-C distance, and Rb (dashed line) corresponding to the (C-Hb) distance for the IRC path of the SN2 reaction. In the side frame: detail of the minima observed for the bond distances at RX ≈ -0.3. Distances in Angstroms.
To support our previous observations, we find it instructive to plot the distances between the incoming hydrogen (Ha) and the leaving hydrogen (Hb) in Fig. 7. These distances exhibit stretching and elongation features associated with bond forming and breaking situations, as anticipated. In contrast to the previously analyzed abstraction reaction, the SN2 reaction occurs in a concerted manner, where bond breaking and forming start simultaneously in a gradual and more intricate manner, as explained below. An interesting feature observable from Fig. 7 is that while the elongation of the carbon-nucleofuge (C-Hb) bond (Rb) changes its curvature significantly at RX ≈ -1.7 (in the forward direction of the reaction), the stretching of the nucleophile-carbon (Ha-C) bond (Ra) occurs smoothly. This suggests that bond breaking occurs first due to the repulsive forces exerted by the ionic molecule as the nucleophile approaches, causing the carbon-nucleofuge bond to break as the molecule begins to release its kinetic energy (resulting in a decrease of momentum entropy). In this sense, the reaction occurs in a concerted manner, with bond breaking and dissipating energy processes occurring simultaneously. In the vicinity of the transition state (TS), around RX ≈ -0.3, we observe small changes in both interatomic distances, as revealed through minima in the amplified picture, indicating the presence of repulsive forces at the TS. Moreover, we analyze the internal angle between Ha-C-H along with the Shannon entropy in position space for comparison purposes. The internal angle clearly indicates that the molecule begins to undergo the "inversion of configuration" at around RX ≈ -1.7, where the nucleophile displaces the nucleofuge from the backside in a single concerted reaction step. This process initiates at the BCER regions, as mentioned above.
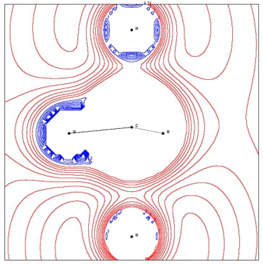
Fig. 8 The MEP contour lines in the plane of Ha-C-Hb (Ha stands for the nucleophilic atom and Hb is the nucleofuge, on bottom and top, respectively) showing positive MEP (nucleophilic regions) and negative MEP (electrophilic regions) at RX ≈ -0.3 for the SN2 reaction.
Fig. 8 illustrates the repulsive effect mentioned earlier in relation to the interatomic distances (Fig. 7), highlighting that the leaving atom Hb gains nucleophilic power (negative MEP). Although not depicted, the transition state (TS) exhibits a half-and-half electrophilic/nucleophilic character among the atoms, with the charge evenly distributed throughout the molecule.
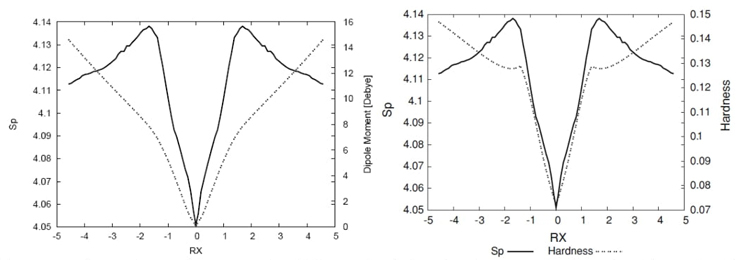
Fig. 9 Shannon entropy in position space (solid line), the total dipole moment values in Debyes (dotted line), and the and the hardness values (dashed line) for the IRC path of the SN2 reaction.
The SN2 reaction serves as an excellent probe to examine the polar
bond pattern characteristic of heterolytic bond-breaking, with residual ionic
attraction due to the ionic nature of the products. This characteristic should
be reflected through the dipole moment of the molecules along the IRC path (note
that the origin of the coordinate system is placed at the molecule's center of
nuclear charge). This is indeed observed in Fig.
9, where these values, along with the ones of the momentum entropy,
are depicted for comparison purposes. At the transition state (TS), the dipole
moment is zero, indicating the nonpolar character of the TS structure, with both
nucleophile and nucleofuge atoms evenly repelling each other through their
carbon bonding. At this point, the momentum and position entropies are minimal
and maximal, respectively, reflecting the low kinetic energy feature of the
chemically relaxed TS structure. As the reactive complex approaches the
reactants/products regions, the dipole moment increases monotonically,
reflecting the polar bonding character of the ionic complex, with a significant
change of curvature at the TS vicinity at around |RX| ≈ 1.0 (a change
of curvature was already noted for all entropies at the same region). In the
transition from reactants to products, the inversion of the dipole moment values
clearly reflects the inversion of configuration of the molecule (this reaction
begins with a tetrahedral
It is interesting to note that as in the case of the hydrogenic abstraction reaction, the eigenvalues of the Hessian [24] for the normal mode associated with the TS along the IRC path show maxima at the BCER and reach their minimal value at the TS. This again validates the concept of a continuum of transient of Zewail and Polanyi , i.e., a transition region rather than a single transition state [38,70].
The effectiveness of theoretic-information measures of the Shannon type in characterizing elementary chemical reactions has been evaluated. The transition state (TS) regions of both chemical reactions were identified, and a plausible connection between the TS and the Shannon entropies was provided and numerically verified. Furthermore, through these chemical probes, we observed basic chemical phenomena of bond breaking and forming, demonstrating that Shannon measures are highly sensitive in detecting these events. Additionally, while the transition state of a reaction is typically identified by the presence of a negative force constant for one normal vibrational mode corresponding to an imaginary frequency, the work of Zewail and Polanyi [70] in transition state spectroscopy has introduced the concept of a reaction having a continuum of transient, a transition region rather than a single transition state. It is noteworthy that the results of the present study indeed demonstrate the existence of such a region between the bond critical energy regions (BCER), before and after the TS. This finding is consistent with reaction force (F(R)) studies, where the reaction force constant (κ(R)) also reflects this continuum, bounded by the minimum and maximum of F(R), at which κ(R) = 0. Results of the study have been reported in Ref. [24].
3D complexity analysis of the hydrogenic abstraction reaction
In recent years, there has been a growing interest in applying complexity concepts to the study of physical, chemical, and biological phenomena. Complexity measures are generally understood as indicators of pattern, structure, and correlation within systems or processes. Several mathematical approaches have been proposed to quantify complexity and information, including Kolmogorov-Chaitin or algorithmic information theory [71], Shannon and Weaver's classical information theory [72], Fisher information [58,73], logical depth [74], and thermodynamical depth [75]. The definition of complexity is not universally agreed upon, and its quantitative characterization has been a significant research focus, receiving considerable attention [76,77]. The utility of each complexity definition depends on the type of system or process under study, the level of description, and the scale of interactions among particles, atoms, molecules, biological systems, etc. Fundamental concepts like uncertainty or randomness are often employed in complexity definitions, but other concepts such as clustering, order, localization, or organization might also play important roles in characterizing system complexity. It is unclear how these concepts should be integrated to quantitatively assess system complexity. However, recent proposals have formulated complexity as a product of two factors, considering order/disequilibrium and delocalization/uncertainty. For instance, the López-Mancini-Calbet (LMC) shape complexity measure [76-79] satisfies boundary conditions by reaching minimal values in extremely ordered and disordered limits. The LMC measure is constructed as the product of two significant information-theoretic quantities: disequilibrium D (also known as self-similarity [80] or information energy [81]), which quantifies the deviation of the probability density from uniformity [79,82] (equiprobability), and Shannon entropy S , which measures randomness/uncertainty in the probability density [72] and quantifies the departure from localizability. Both global quantities are closely related to the spread of a probability distribution.
The Fisher-Shannon product (FS) has been used as a measure of atomic correlation [83] and defined as a statistical complexity measure [84]. The product of power entropy J -explicitly defined in terms of Shannon entropy-and Fisher information measure I combines global characteristics (depending on the distribution as a whole) with local characteristics (related to the gradient of the distribution), preserving general complexity properties. Unlike LMC complexity, which relies on disequilibrium to measure global deviation from uniformity, Fisher-Shannon complexity uses Fisher information to quantify local distribution disorder [73,85].
We have undertaken an information-theoretical complexity study of the hydrogenic abstraction reaction [29] by use of information-theoretical measures and planes as well as the LMC and FS complexity products. The recognition of patterns of uncertainty/localizability, disorder/narrowness and disequilibrium/uniformity, were characterized, through the S, I and D functionals, respectively.
Complexities
The LMC complexity is defined through the dyadic product of information-theoretic measures. So that, in position space, the probability density ρ(r), is employed to obtain the C(LMC) complexity [76-79]:
where D r is the disequilibrium information measure [80,81]
and S is the Shannon entropy [72], Eq (1):
from which the exponential entropy
It is important to mention that the LMC complexity of a system must comply with the following lower bound [84]:
The FS complexity in position space, C r (FS), and similarly in momentum space, C p (FS), are defined in terms of the product of Fisher information I [73,85] and the power entropy [86], J, (See below),:
in position space and
in momentum space, where ρ(r) and γ(p) denote the normalize-to-unity electron density distributions in the position and momentum spaces, respectively. The power entropy [86] in position space, J r , is defined as
and similarly for J p in momentum space. Both depending on the Shannon entropy defined in Eq. (1). So that, the FS complexity in position space is given by
and similarly
in momentum space.
Let us remark that the factors in the power Shannon entropy J are chosen to preserve the invariance under scaling transformations, as well as the rigorous relationship [87].
with n being the space dimensionality, thus providing a universal lower bound to FS complexity. The definition in Eq. (18) corresponds to the particular case n=3, the exponent containing a factor 2/n for arbitrary dimensionality.
Note that the inequalities above remain valid for distributions normalized to unity, which is the choice that it is employed throughout this work for the 3-dimensional molecular case.
Aside of the analysis of the position and momentum information measures, we have
defined these magnitudes in the product rp-space, characterized
by the probability density
And
From the above two equations, it is evident that the features and patterns of both LMC and FS complexity measures in the product space will be determined by those of each conjugated space. However, the numerical analyses conducted in the next section reveal that the momentum space contribution plays a more significant role compared to the one in position space.
3D complexity analysis of the hydrogenic abstraction reaction
The electronic structure calculations performed in the present study were carried out with the Gaussian 03 suite of programs [62]. Reported TS geometrical parameters for the abstraction reaction were employed [64]. Calculations for the IRC were performed at the MP2 (UMP2 for the abstraction reaction) level of theory with at least 35 points for each one of the directions (forward/reverse) of the IRC. Next, a high level of theory and a well-balanced basis set (diffuse and polarized orbitals) were chosen for determining all of the properties for the chemical structures corresponding to the IRC. Thus, the QCISD(T) method was employed in addition to the 6-311++G** basis set, unless otherwise stated. The molecular information measures S, D, I, J; the information planes (D-L), (I-J), (I-D) and the complexity measures, C(LMC) and C(FS). All information-theoretical quantities are calculated in position and momentum spaces for the IRC path of the abstraction reaction and obtained by employing software developed in our laboratory along with 3D numerical integration routines [66], and the DGRID suite of programs [67]. The IRC represents a minimum energy reaction pathway (MERP) resembling a chemical road crossing through a 3D horse saddle, forming an energy hypersurface. We constructed such a 3D-saddle surface by extending the internal coordinates of the three-hydrogen complex Ha ··· H b ··· H c in all directions using an arbitrary equidistant grid ranging from 00.5a.u. ≤ R 12 ≤ 3.35a.u. vs. 0.5a.u. ≤ R 13 ≤ 3.35 a.u. in steps of 0.05 a.u., far beyond the IRC. Once the grid was formed, a 3D information-theoretical analysis of the hydrogenic abstraction reaction H • a + H 2 ⇋ H 2 + H • was conducted using information-theoretical measures and the dyadic products of statistical complexity (LMC and FS). The focus was set on recognizing 3D patterns of localizability, disorder, and uniformity through the hypersurfaces of S, I, and D, respectively. This is achieved by calculating the 3D surfaces of the IT components, along with the Fisher-Shannon and LMC complexities, in both position ( r ) and momentum ( p ) spaces. The diagonal path of the IT-functionals hypersurfaces will be examined in light of the equidistant dissociation of the three-atomic complex molecule at the TS H a ··· H b ··· H c .
Understanding the structural characteristics of distributions in both position and momentum spaces, particularly in terms of global spreading (delocalization) of densities, can be achieved through Shannon entropies in conjugated spaces. However, Fisher information is more suitable for describing the behavior of densities concerning their local changes [73,85]. This measure evaluates the pointwise concentration of the electronic probability cloud in each space by examining the gradient of the electron distribution. This allows it to reveal changes in density and provide a quantitative assessment of its oscillatory nature (smoothness).
It's crucial to note that the global or local nature of these information measures means that each can only partially describe the complete chemical phenomena. These phenomena include identifying regions such as reactant/product complexes (R/P), bond breaking/forming regions (B-B/F), bond cleavage energy reservoirs (BCER), transition states (TS), and mechanistic behaviors. Therefore, to fully understand a chemical process, a complexity analysis is essential. These information-theoretical measures offer complementary perspectives: disequilibrium (D) quantifies deviations from uniformity, while Shannon entropy (L) measures deviations from localizability, as described by the C(LMC) complexity measure. In a similar vein, Fisher information (I) assesses deviations from disorder, and power entropy (J) evaluates deviations from localizability, forming the basis of the C(FS) measure (Eqs. 8, 11, 16, and 17).
In Fig. 9, we have depicted the LMC complexity surface in 3D in position space as a function of the internal coordinates R 12 and R 13. A top view of the hypersurface of the C LMC complexity measure in the X-Y plane is also shown. Similarly, Figure 10 presents the C FS complexity measure, both figures indicating the IRC pathway of the reaction along with the chemically significant zones: R/P, B-B/F, BCER, and TS. The color codes represent lower to higher complexity values, ranging from bluish to yellowish.
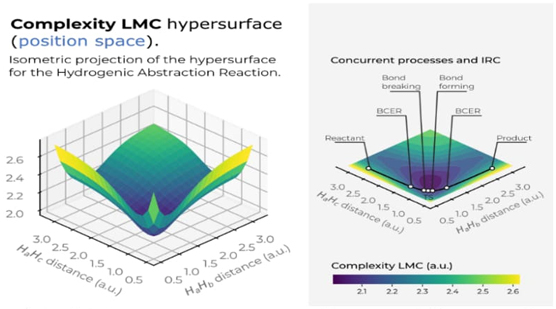
Fig. 9 Hypersurface of the LMC complexity measure in position space (left) and top view of its 3D surface in the X−Y plane (right) for the hydrogenic abstraction reaction in the grid of the internal coordinates, R 12 and R 13, of the three hydrogenic complex H a ··· H b ··· H c . Color codes indicate lower to higher LMC-complexity values running from yellowish to bluish ones, respectively.
Fig. 9 displays a deep narrow well that encompasses all the crucial regions of the reaction, where concurrent phenomena occur, such as the B-B/F, BCER, and TS regions. This suggests that significant chemical changes happen in regions of lower LMC complexity (greenish areas in Fig. 9). It would be intriguing to further explore the chemical implications of this low-complexity well, which might indicate a boundary delimiting structurally stable molecules. Notably, Fisher's hypersurface (Fig. 5 in Ref [90]) reveals a similar region, despite being a different type of functional compared to the global ones used to represent C LMC This indicates that both local and global IT functionals detect this low-complexity boundary. Therefore, the deep narrow well likely represents a chemically significant boundary. Further, the 3D surface of C LMC indicates that higher LMC-complexity regions are noticed at the dissociated complex species, i.e., (H a ··· H b ··· H c )· → H 2 + H · c at very large R bc , (H a ··· H b ··· H c )· → H · a + H 2 at very large R ab , and (H a ··· H b ··· H c )· → (H a + H b + H c )· at very large R aBocbc . This observation allow us to conclude the higher complex behavior of the dissociated species.
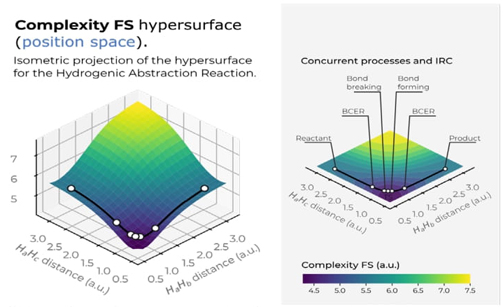
Fig. 10 Hypersurface of the Fisher−Shannon complexity measure in position space (left) and top view of its 3D surface in the X−Y plane (right) for the hydrogenic abstraction reaction in the grid of the internal coordinates, R 12 and R 13, of the three hydrogenic complex H a ··· H b ··· H c . Color codes indicate lower to higher Fisher−Shannon complexity values running from yellowish to bluish ones, respectively.
Fig. 10 illustrates a deep narrow slope at small distances of the internal coordinates R 12 and R 13, signifying a low Fisher-Shannon complexity C FS region. This region interestingly also encloses the chemically significant zones where concurrent phenomena occur, such as B-B/F, BCER, and TS, similarly to the LMC complexity depicted in Fig. 9. Additionally, higher Fisher-Shannon complexity regions are found in the dissociation zone of this complex radical molecule: (H a ··· H b ··· H c )· → (H a + H b + H c )· at very large R abc , which contrasts with the LMC complexity that shows higher values for all types of dissociated species. The boundaries of low-complexity regions for both measures, C LMC and C FS , enclose complex radical molecules with lower complexity values than the TS, as seen in Figures 9 and 10. This observation is chemically significant and warrants further investigation. Furthermore, an interesting observation from these figures is that the hypersurfaces for both complexities in position space fold around the TS region, revealing an attractor-like spatial zone.
The combined analyses of the 3D structure of the information-theoretic (IT) functionals demonstrate that the chemically significant regions at the onset of the TS are thoroughly characterized by aspects of localizability (S), uniformity (D), and disorder (I) . Additionally, novel regions of low complexity suggest new boundaries for chemically stable complex molecules. The study further reveals that the chemical reaction occurs in low-complexity regions where concurrent phenomena such as bond-breaking/forming (B-B/F), bond-cleavage energy reservoirs (BCER), spin-coupling (SC), and the transition state (TS) take place. The complexity measures in both spaces display folded hypersurfaces around the TS region, indicating attractor-like spatial/momentum zones. Moreover, the focus has been on the diagonal part of the hypersurface of the IT functionals, beyond the IRC path itself, to analyze the dissociation process of the triatomic transition-state complex, revealing additional interesting features of the bond-breaking (B-B) processThe material presented in this Section has been recently published (Ref. [90]).
Atoms and molecules
The Fisher-Shannon and LMC shape complexities, along with the Shannon-disequilibrium, Fisher-Shannon, and Fisher-disequilibrium information planes, which comprise two localization-delocalization factors, have been recently computed in both position and momentum spaces for the one-particle densities of 90 selected molecules of various chemical types [91]. It was found that while the two measures of complexity show only general trends, the localization-delocalization planes exhibit distinct chemically significant patterns. Several molecular properties (energy, ionization potential, total dipole moment, hardness, electrophilicity) were analyzed to interpret and understand the chemical nature of the composite information-theoretic measures. The results indicate that these measures can detect not only randomness or localization but also pattern and organization.
For larger molecules, Shannon Information Theory (IT) has been applied to analyze the growth behavior of nanostructures [92-94].
Some other topics of interest have been included in this part of the Review: (i) predominant information quality schemes (PIQS) of amino acids [95] , (ii) 3D information-theoretic analyses of the chemical space from simple atomic and molecular systems to biological and pharmacological molecules [96], and (iii) the separability problem of atoms in molecules [97].
Complexity and information plans of selected molecules
Following the complexity concepts discussed above, we conducted an information-theoretical analysis of ninety molecular systems of various chemical types to analyze and quantify their information content [91]. The focus was on recognizing patterns of uncertainty, order, and organization by employing several molecular properties such as energy, ionization potential, hardness, and electrophilicity. We computed the information components, as well as the Fisher-Shannon and LMC complexities, C(FS) and C(LMC), respectively (Eqs 8 and 16). These information functionals of the one-particle density were computed in position ( r ) and momentum ( p ) spaces, as well as in a joint product space ( rp ) that contains more comprehensive information about the system. Eqs. (19) and (20). Additionally, the Fisher-Shannon (I-J) and the disequilibrium-Shannon (D-L) planes were studied to identify patterns or organization.
These complexity measures and their associated informational planes were analyzed in terms of their chemical properties, number of electrons, and geometrical features. The molecular set chosen for this study includes a variety of chemical organic and inorganic systems, such as aliphatic compounds, hydrocarbons, aromatics, alcohols, ethers, and ketones. This set represents a range of closed-shell systems, radicals, isomers, and molecules containing heavy atoms such as sulfur, chlorine, magnesium, and phosphorus. The geometries needed for the single-point energy calculations were obtained from the Computational Chemistry Comparison and Benchmark Database from NIST [98]. The molecular set can be organized into isoelectronic groups as follows:
N-10: NH 3 (ammonia),
N-12: LiOH (lithium hydroxide),
N-14: HBO (boron hydride oxide), Li 2 O (dilithium oxide),
N-15: HCO (formyl radical), NO (nitric oxide),
N-16; H 2 CO (formaldehyde), NHO (nitrosyl hydride), O 2 (oxygen),
N-17: CH 3 O (methoxy radical),
N-18: CH 3 NH 2 (methyl amine), CH 3 OH (methyl alcohol), H 2 O 2 (hydrogen peroxide), NH 2 OH (hydroxylamine),
N-20: NaOH (sodium hydroxide),
N-21: BO 2 (boron dioxide), C 3 H 3 (radical propargyl), MgOH (magnesium hydroxide), HCCO (ketenyl radical),
N-22: C 3 H 4 (cyclopropene), CH 2 CCH 2 (allene), CH 3 CCH (propyne), CH 2 NN (diazomethane), CH 2 CO (ketene), CH 3 CN (acetonitrile), CH 3 NC (methyl isocyanide), CO 2 (carbon dioxide), FCN (cyanogen fluoride), HBS (hydrogen boron sulfide), HCCOH (ethynol), HCNO (fulminic acid), HN 3 (hydrogen azide), HNCO (isocyanic acid), HOCN (cyanic acid), N 2 O (nitrous oxide), NH 2 CN (cyanamide),
N-23: NO 2 (nitrogen dioxide), NS (mononitrogen monosulfide), PO (phosphorus monoxide), C 3 H 5 (allyl radical), CH 3 CO (acetyl radical),
N-24: C 2 H 4 O (ethylene oxide), C 2 H 5 N (aziridine), C 3 H 6 (cyclopropane), CF 2 (difluoromethylene), CH 2 O 2 (dioxirane), CH 3 CHO (acetaldehyde), CHONH 2 (formamide), FNO (nitrosyl fluoride), H 2 CS (thioformaldehyde), HCOOH (formic acid), HNO 2 (nitrous acid) NHCHNH 2 (aminomethanimine), O3 (ozone), SO (sulfur monoxide),
N.25: CH 2 CH 2 CH 3 (npropyl radical), CH 3 CHCH 3 (isopropyl radical), CH 3 OO (methylperoxy radical), FO 2 (dioxygen monofluoride), NF 2 (difluoroamino radical), CH 3 CHOH (ethoxy radical), CH 3 S (thiomethoxy),
N-26: C 3 H 8 (propane), CH 3 CH 2 NH 2 (ethylamine), CH 3 CH 2 OH (ethanol), CH 3 NHCH 3 ( dimethylamine), CH 3 OCH 3 (dimethyl ether), CH 3 OOH (methyl peroxide), F 2 O (difluorine monoxide),
N-30: ClCN (chlorocyanogen), OCS (carbonyl sulfide), SiO 2 (silicon dioxide),
N-31: PO 2 (phosphorus dioxide), PS (phosphorus sulfide),
N-32: ClNO (nitrosyl chloride), S 2 (sulfur diatomic), SO 2 (sulfur dioxide),
N-33: OClO (chlorine dioxide), ClO 2 (chlorine dioxide),
N-34: CH 3 CH 2 SH (ethanethiol), CH 3 SCH 3 (dimethyl sulfide), H 2 S 2 (hydrogen sulfide), SF 2 (sulfur difluoride),
N-38: CS 2 (carbon disulfide),
N-40: CCl 2 (dichloromethylene), S 2 O (disulfur monoxide),
N-46: MgCl 2 (magnesium dichloride),
N-48: S 3 (sulfur trimer), SiCl 2 (dichlorosilylene),
N-49: ClS 2 (sulfur chloride).
The electronic structure calculations performed in the present study for the whole set of molecules were obtained by use of correlated wavefunctions at high levels of theory.
Complexity measures
In contrast to the atomic case, where complexities exhibit a high level of natural organization due to periodicity properties [99,100], the molecular case necessitates some form of organization or classification, which can be influenced by various factors such as structural, energetic, and entropic characteristics. Therefore, we analyzed the molecular complexities, C(LMC) and C(FS), as functions of key chemical properties of interest, including total energy, dipole moment, ionization potential, hardness, and electrophilicity. By establishing a link between the different complexity measures and these chemical properties, we aimed to gain insights into the organization, order, and uncertainty features of the molecular systems.
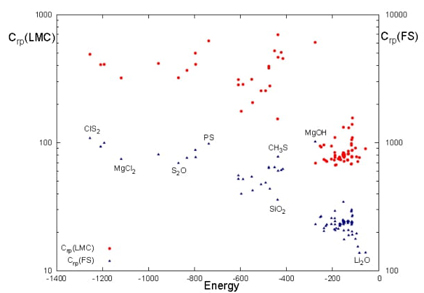
Fig. 11 C(LMC) (red circles) and C(FS) (blue triangles) complexities as a function of the total energy (a.u.) for the set of ninety molecules in the product space (rp).
In Fig. 11, we have depicted the
C(FS) and C(LMC) complexities as functions
of the total molecular energy (in atomic units) within the product space (rp).
Observations from the figure indicate that both complexity measures show similar
patterns. A noticeable trend is that molecules with higher energies tend to
exhibit increased complexity values for both C(FS) and
C(LMC), compared to those with lower energies. The
molecules analyzed are categorized into four energy intervals: E > - 400, E
These findings suggest that molecular complexity is influenced by several factors beyond energy, including molecular structure, composition, and chemical functionality, which contribute to varying degrees of complexity. For example, the lowest complexity values within each energy group typically align with molecules that share similar geometrical configurations, while higher complexities are found in molecules containing heavier atoms. Each complexity measure integrates two components: one invariably linked to Shannon entropy S , and the other differentiates the measures-disequilibrium D for C rp (LMC) and Fisher information I for C rp (FS), representing global and local perspectives, respectively. Despite these distinctions, no significant structural differences are evident between the complexities determined by D or I .
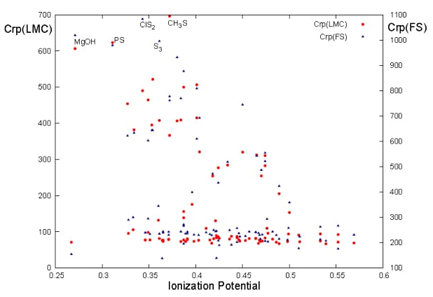
Fig. 12 C(LMC) (red circles) and C(FS) (blue triangles) complexities as a function of the ionization potential (a.u.) for the set of ninety molecules in the product space (rp).
Ionization potential (IP) is utilized to gauge the chemical stability of molecules in relation to their complexities. As depicted in Fig. 12, molecules with higher IP values, indicating greater stability, appear on the right side of the figure. This observation highlights a correlation between molecular complexities and stability: higher LMC and FS complexities are associated with molecules that are more reactive and, consequently, less stable.
Information planes
In our search for patterns and organization, we have found it useful to analyze the molecules in our study based on their energy and the number of electrons. This analysis involves plotting the contribution of information measures D (order) and L (uncertainty) to the total complexity of the LMC, as well as measures I (organization) and J (uncertainty) to the FS complexity. Figures 13 and 14 show the behavior of energy in the D p -L p plane and the effect of the number of electrons in the I p -J p planes, respectively. When analyzing the energy in the information planes, we have found it more beneficial to focus on the corresponding planes in momentum space, as the momentum density is directly linked to energy. In our publication (Ref. [91]), we presented results pertaining to the behavior of energy in the I p -J p planes and the influence of the number of electrons in the D r -L r planes.
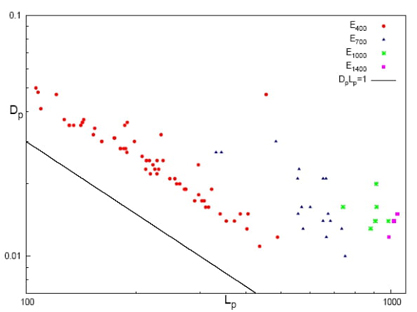
Fig. 13 Disequilibrium-Shannon plane (D-L) in momentum
space for energetically differents groups: E400 for
molecules with E > - 400 (red cicles),
E700 for E
In Fig. 13, the D
p
-L
p
plane is shown in a double-logarithmic scale for a set of molecules
grouped according to their energy intervals. The energy intervals are labeled as
E400, E700, E1000, and E1400, corresponding to different ranges of energy, i.e.,
E400 for molecules with E > -400 a.u.,
E700 for E
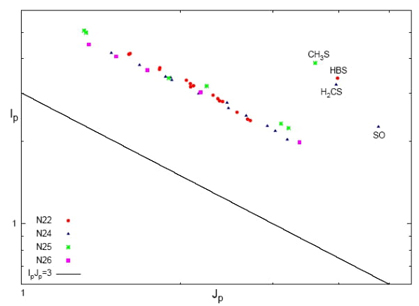
Fig. 14 Fisher-Shannon plane (I-J) in momentum space of the isoelectronic series of 22 (red cicles), 24 (blue triangles), 25 (green stars), and 26 (magenta box) electrons. Double-logarithmic scale. Lower bound (I p -J p =3) is depicted by the black line. Molecules with larger energy vaues are shown at the upper left corner of the Figure.
In the analysis of pattern and organization for the isoelectronic series, we have examined how different information measures contribute to the overall complexity of the system. Fig. 14 displays this analysis for a selection of isoelectronic molecular series with varying numbers of electrons in the momentum space: N = 22, 24, 25 and 26 electrons in the I p -J p plane. The parallel lines on the graph represent isocomplexity lines, indicating regions of equal complexity. Deviations from these lines indicate higher levels of complexity. When there is an increase in uncertainty ( J ), there is typically a corresponding decrease in accuracy (I ) along these lines. This linear relationship is observed consistently across all isoelectronic series in momentum space, as illustrated in Fig. 14. The accuracy and uncertainty values were determined through linear regression analysis, yielding high correlation coefficients for each series (N22: 0.989, N24: 0.994, N25: 0.993, N26: 0.998). It is important to note that systems outside the isocomplexity lines belong to molecules with higher complexity, as discussed earlier. These systems typically contain heavier atoms, as indicated in Figure 14, leading to higher uncertainty values (J p ) and therefore greater complexity. Furthermore, the isocomplexity lines representing the same isoelectronic molecular series exhibit significant deviations (higher complexity) from the strict lower bound, as shown in the figure. In the conjugated position space (I r -J r ), a similar trend is observed. Each isoelectronic series demonstrates a linear relationship, with exceptions for molecules with the highest complexity values that fall outside the isocomplexity lines. These complex molecules have lower values of J r and higher values of I r in contrast to the behavior observed in Fig. 14.
Despite the limitations of using all information products as complexity measures, such as the requirement of invariance under scaling, translation, and replication, we found it intriguing to examine patterns of order and organization in the I-D plane. Our analysis of these patterns is detailed in Ref. [91].
In our analysis, we found that fulminic acid (HCNO) has the lowest ionization potential compared to isocyanic acid (HNCO) and cyanic acid (HOCN). This indicates that this acid is less stable and more reactive than the other two isomers. This observation is consistent with the complexity values obtained from our analysis. From the Table 1 of chemical properties (below), we can see that fulminic acid has larger values for the complexity measures compared to isocyanic acid and cyanic acid. This suggests that fulminic acid possesses more intricate patterns of order and organization in its molecular structure. The higher complexity values indicate a higher level of complexity and structural diversity in this molecule ¡Error! No se encuentra el origen de la referencia.. Based on our previous discussion regarding the relationship between complexity measures and chemical properties, a molecule with higher complexity values is expected to be more reactive. In this case, fulminic acid, being the isomer with the highest complexity measures, aligns with this expectation and can be considered as a more reactive molecule. Overall, the analysis of the complexity values for these isoelectronic isomers provides insights into their chemical properties and corroborates the experimental knowledge regarding their stability and reactivity.
Table 1 Chemical properties and complexity measures for the isomers HCNO, HNCO and HOCN in atomic units (a.u.).
| Molecule | Energy | Ionization Potential | Hardness | Electrophilicity | Crp(LMC) | Crp(FS) |
|---|---|---|---|---|---|---|
| HCNO | -168.134 | 0.403 | 0.294 | 0.020 | 76.453 | 229.303 |
| HNCO | -168.261 | 0.447 | 0.308 | 0.031 | 74.159 | 223.889 |
| HOCN | -168.223 | 0.453 | 0.305 | 0.036 | 75.922 | 225.656 |
Our analyses show that molecular complexity is influenced by several chemical factors, including molecular structure, composition, functionality, and reactivity. Molecules with higher complexity values tend to have higher energies, larger numbers of electrons, smaller hardness values (indicating higher reactivity), and smaller ionization potential values (indicating lower stability). However, there is no clear correlation between molecular complexity and chemical reactivity, suggesting that other factors beyond complexity also play a role in determining reactivity. Furthermore, the analysis of information planes reveals that molecular energies are related to the uncertainty of the systems, as measured by L p or J p . Low-energy molecules tend to exhibit more linear behavior and are closer to the lower bounds, whereas high-energy molecules tend to deviate from the iso-ocomplexity lines. This suggests that complexity is influenced by both molecular energy and uncertainty. In summary, molecular complexity is affected by a combination of factors including structure, composition, reactivity, and energy. The analysis of information planes provides further insights into the relationship between complexity and uncertainty in molecular systems.
Predominant information-theoretic quality scheme (PIQS) of amino acids
To our knowledge, no prior studies have thoroughly characterized essential amino acids using information-theoretic measures of their actual electronic densities. In this research, we analyzed probability density distributions derived from wave-function calculations at the Hartree-Fock (HF) and Configuration Interaction (CI) levels of theory. We computed several metrics: Shannon entropy, Fisher information, disequilibrium, and the FS and LMC complexities to categorize amino acids based on their physicochemical traits. The objectives of this study are multifaceted: firstly, to describe amino acids through their inherent information content, utilizing measures that reflect delocalization, narrowness, and order; secondly, to develop a comprehensive classification system for amino acids based on their biochemical properties; and thirdly, to explore potential connections between information measures and reactivity parameters. Results have been published in Ref. [95].
Computational details
In this study, we performed electronic structure calculations for a complete set of amino acids using the Gaussian 09 software suite [62] at the Hartree-Fock (HF), DFT-M062X, and CISD(Full) levels of theory with the 6-311+G(d,p) basis set. We initially conducted geometry optimizations on 18 amino acids extracted from the 1C3X protein, considering five distinct conformations for each amino acid (excluding cysteine and histidine, which are absent from this protein). To maintain the integrity of the original biological context, we restricted our optimizations to only the hydrogen atoms while preserving the skeletal structure of each conformation. These optimizations were carried out at the HF level using the specified basis set. Subsequent single-point calculations at the CISD(Full) level were then performed to determine all the information-theoretic properties relevant to our analysis. It should be noted that the selection of only five conformers for each amino acid in this study is arbitrary but serves to evaluate the significance of geometric conformations within the protein. The primary aim of this research is to compute information-theoretic measures in position space, which were determined at the CI level.
Bacteriorhodopsin, sourced from the Protein Data Bank of Cambridge University with the identifier PDB ID:1C3W [102], serves as a key subject for study due to its vital role in photosynthesis within Halobacterium salinarum, a type of archaeon. This protein is primarily responsible for converting energy from green light, within the 500-650 nm spectrum, into an electrochemical proton gradient that facilitates ATP synthesis via ATP synthase. Functioning as a light-driven proton pump, it actively transports protons outside of the cell. Bacteriorhodopsin is composed of over 250 amino acids, which include 18 different essential types. Our study particularly focuses on analyzing various conformations of these amino acids-specifically examining different positions within the protein to understand their behavior in a natural biological context. It is crucial to note that the structural properties of an amino acid are significantly influenced by its environmental conditions, which include the potential interactions with neighboring amino acids and spatial interactions within its vicinity. Therefore, the physicochemical properties of amino acids are influenced by these environmental interactions, suggesting that a single property might not adequately represent an amino acid's characteristics due to its conformational diversity within the protein structure (see for instance Fig.15). However, we propose that information-theoretic measures could be more effective in characterizing amino acids within proteins, despite their diverse conformations. This is because such measures effectively capture the fundamental aspects of the electron densities in these systems.
Fig. 15 View of the different conformational structures of methionine at residues met-118, met-209, met-20, met-32 and met-60 for 1C3W.
In Fig. 15 the five conformers of methionine are shown through the residues met-118, met-209, met-20, met-32 and met-60.
Amino acids classification: predominant information quality scheme
Several methods have been used to categorize amino acids based on various chemical and biological properties such as hydrophobicity/hydrophilicity, charge/neutrality, acidity/basicity, aliphaticity/aromaticity, and polarity/non-polarity. Additionally, amino acids are classified according to their biological roles, differentiating between proteic (essential, chemically modified) and non-proteic types (d-amino acids, alpha-amino acids, omega-amino acids). Fig. 16 illustrates a Venn diagram that groups amino acids based on these properties [103, 104]. It's challenging to pinpoint physicochemical properties that effectively capture similarities among amino acids, as the substitution of one amino acid for another is more likely to be successful when the two share similar properties. The Venn diagram facilitates the chemical classification of amino acids by grouping those with similar structures into common regions. This visual representation highlights the shared properties and characteristics among different sets of amino acids [104].
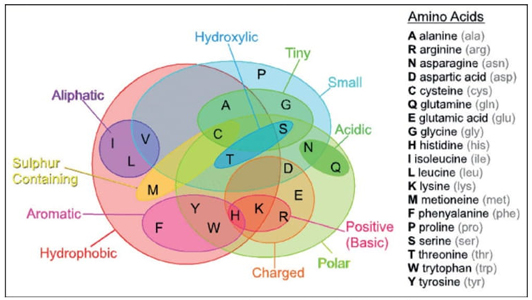
Fig. 17 presents histograms for various properties measured across all conformations of amino acids in the 1C3W protein. These properties include Energy (E), Chemical Potential (µ), Hardness (η), and information-theoretic measures like Shannon entropy (S), Fisher information (I), and Disequilibrium (D). Notably, properties like chemical potential and hardness exhibit considerable variability across different conformations, highlighted in green and yellow, reflecting their strong dependence on the conformeric geometry. In contrast, measures such as energy and information-theoretic values are relatively consistent across different conformations of each amino acid, displayed in red and blue. This consistency, particularly in information-theoretic measures, provides a robust characterization of each amino acid, unlike chemical potential where the values for amino acids like lysine (K) and phenylalanine (F) are indistinguishable. Additionally, other DFT properties like softness, ionization potential, and electrophilicity, not depicted in the histogram, also show significant variability.
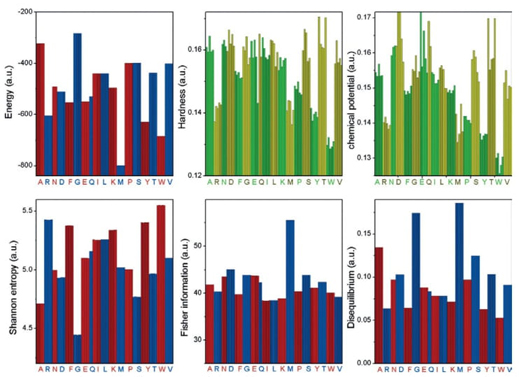
Fig. 17 Energy, hardness, chemical potential, Shannon entropy, Fisher and disequilibrium for the conformational structures of all amino acids present in protein. Figures indicated in red and blue represent the information-theoretic measures and energy for different amino acids, and figures indicated in green and yellow represent DFT reactivity parameters for different amino acids.
Building on the foundational aspects of Information Theory (IT) we've discussed, it's clear that three distinct features-delocalization ( S ), uniformity ( D ), and narrowness ( I )- can be illuminated through information-theoretic measures. These characteristics are inherently present in all chemical structures and are reflected in their electronic densities, which manifest through Shannon entropy, disequilibrium, and Fisher information, respectively. This foundation leads us to propose that all amino acids can be uniquely characterized by their information content, highlighting one of these three distinct IT qualities that differentiate them from other chemical structures. This proposal is supported by data presented in Fig. 18, which shows the average values of these three information measures for each amino acid, calculated across all conformeric structures. The small variance in these values, as noted in Fig. 17, supports the reliability of using averaged data to characterize each amino acid effectively.
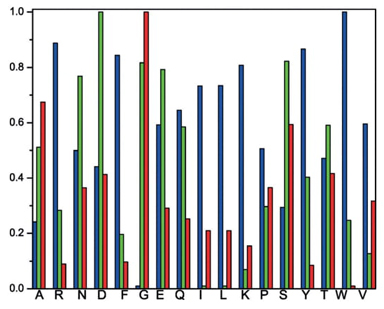
Fig. 18 Average values (of all conformeric structures for each amino acid) for Shannon entropy (blue), Fisher information (green) and disequilibrium (red).
Based on this methodology, we can categorize the 18 essential amino acids using the framework of information qualities, as illustrated in Fig. 19. Each column in this figure groups different amino acid structures according to their dominant information-theoretic qualities: delocalization (S), uniformity (D), and narrowness (I). These are arranged to reflect the significance of each IT feature within a given triplet by order of importance. For instance, in the first column, labeled as SID , the amino acids listed prioritize Shannon entropy as the most significant IT quality, followed by disequilibrium and Fisher information, highlighting a sequence of importance from highest to lowest.
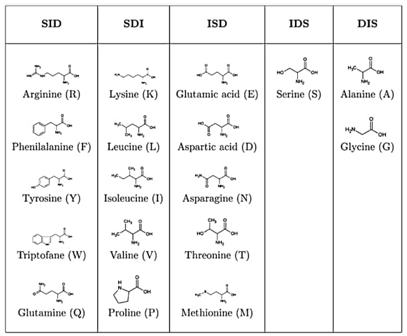
Fig. 19 Classification of amino acids through a predominance information quality scheme (PIQS): Shannon entropy (S), Fisher information (I) and disequilibrium (D).
The study reveals that each amino acid is distinctly characterized by a Predominance Information Quality Scheme (PIQS), which utilizes three fundamental information-theoretic attributes: delocalization, narrowness, and uniformity. This novel classification identifies four primary chemical families: aliphatic (characterized by delocalization), aromatic (also delocalized), electro-attractive (defined by narrowness), and tiny (marked by uniformity). This grouping enhances and integrates existing classifications such as sulfur-containing, hydrophobic, hydroxylic, charged, basic, polar, small, and tiny amino acids.
Additionally, the study highlights the consistency of these classifications through diadic products of information-theoretic measures, reinforcing the PIQS. Importantly, the analysis also shows that the information-theoretic measures and energy levels exhibit a consistent pattern across different conformeric structures of the same amino acid within various residues of the protein, indicating a stable profile for these measures. Conversely, other chemical properties like softness, ionization potential, chemical potential, and electrophilicity display significant variability across the study, underlining their dependency on specific amino acid conformations. Results of the study have been reported in Ref. [95].
3D-Information-theoretic chemical space from atoms to molecules
In this subsection, we introduce a three-dimensional Information-Theoretic Space, crafted from Shannon entropy, Fisher information, and disequilibrium, alongside their corresponding Fisher-Shannon and LMC complexity measures. This framework uniquely highlights the distinct physical, chemical, and biological characteristics of a broad array of many-electron systems. These range from atomic systems, both neutral and ionized, to simple molecules and more complex entities like amino acids and pharmacological compounds. This advancement is rooted in the fundamental information-theoretic concepts of delocalization, order, uniformity, and complexity. It suggests the possibility of a universal information-theoretic chemical space that could encapsulate all systems in nature.
IT-3D chemical space
There has been significant interest across Physics, Chemistry, and Biology in characterizing and classifying different physical systems based on a few fundamental properties. This task involves the concept of "chemical space," which, while often mentioned, is not clearly defined yet [105, 106]. This space serves two purposes: it organizes a broad array of molecules, such as all organic molecules in biological systems, and functions as a multidimensional descriptor space used in chemoinformatics [107] and for predicting chemical properties from alchemical derivatives [108]. However, the vast number of physicochemical properties available as descriptors is a major challenge. This abundance increases the risk of using irrelevant or redundant descriptors, which can result in different systems being inaccurately grouped together in the same space, complicating their differentiation.
In this study, we introduce a novel framework rooted in information theory (IT) to develop a comprehensive information-theoretic space. This conceptual space is designed to encompass a broad range of atomic and molecular systems, extending from basic atoms and molecules to complex biological and pharmacological entities.
The novelty of our research lies in demonstrating how the properties of a broad spectrum of systems, from simple physical entities to intricate biological ensembles, are intricately linked to three principal informational qualities: spatial distribution, uniformity, and local fluctuations. These elements-delocalization, uniformity, and order-are quantitatively assessed using information-theoretic measures such as Shannon entropy, disequilibrium, and Fisher information, which relate directly to the electron density characteristics of these systems.
Building on this theoretical foundation, our approach proposes that all systems can be distinctly characterized by their information content, reflecting unique combinations of these three informational attributes. We have developed a three-dimensional Information-Theoretic Space (IT-3D), structured along the axes of disequilibrium ( D ), Shannon entropy ( S ), and Fisher information ( I ). To capture more nuanced aspects of system complexity, this framework incorporates two additional dimensions-Fisher-Shannon (FS) and López-Mancini-Calbet (LMC) complexity measures-enhancing our ability to depict and analyze the composite electronic structures across diverse systems. The core findings of this study are presented in Figures 20 to 22, which display the Information-Theoretic Space (IT-3D) encompassing a broad array of systems including physical, chemical, biological, and pharmacological entities, totaling 388 atoms and 115 molecules.
IT-3D atomic space
Fig. 20 illustrates the IT-3D space positioning for 103 neutral atoms, 96 ions (both positively and negatively charged) ranging from helium to cesium, and 9 isoelectronic series extending from helium to neon. Fig. 21 highlights the entropic features within the IT-3D framework, alongside Fisher-Shannon (FS) and López-Mancini-Calbet (LMC) complexity measures for these neutral atoms.
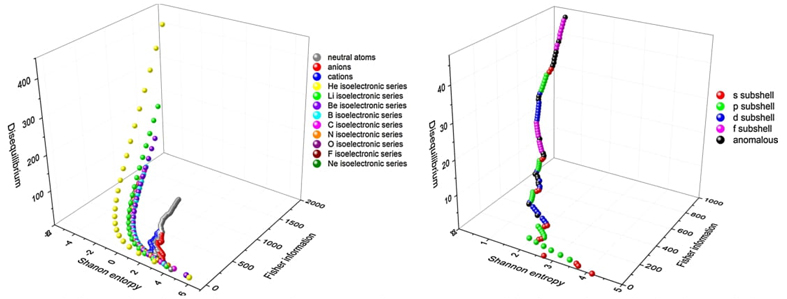
Fig. 20 3D-information-theoretic space of 103 neutral atoms, 96 (positively and negatively charged) ions from He to Cs, and 9 isoelectronic series from He through Ne (left). 3D-IT space for neutral atoms with valence shell filling. Isoelectronic series from N = 2 through N = 10 are depicted with balls on yellow, green, purple, respectively. Neutral atoms and positively and negatively charged ions are depicted by grey, blue and red balls, respectively (right).
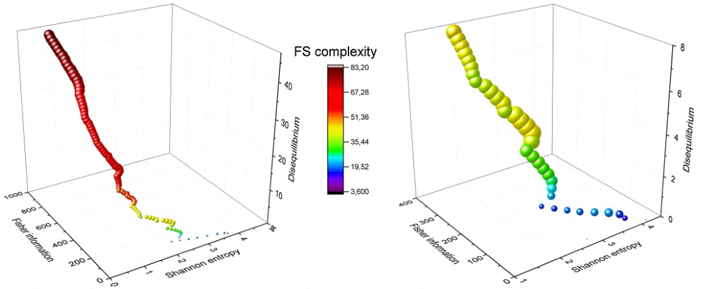
Fig. 21 3D-information-theoretic space for neutral atoms. Higher to smaller values of CFS are depicted in reddish to blueish colours. Similarly, higher to lower values of CLMC are drawn with bigger to smaller ball sizes (left). Detail of the previous graph for the sequence of lighter atoms with N = 2 to N = 35 (right).
IT-3D molecular space
Fig. 22 extends this analysis to a diverse array of complex molecular systems, including 10 alkanes, 4 ethoxides, 52 organic molecules with various bonding types (C-H, C-O, C-N, C-S, C-X where X represents halogens), 20 amino acids, and 29 pharmacologically relevant molecules (encompassing anti-cancer agents, bactericides, antibiotics, anti-inflammatory drugs, and anti-chagas medications). These systems populate distinct regions of the IT-3D space, reflecting their specific information content, which is intrinsically linked to their physicochemical properties. This paper aims to elucidate the fundamental characteristics and significance of this space for the systems detailed above.
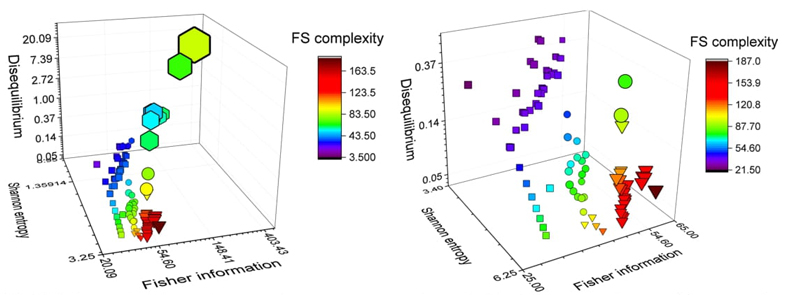
Fig. 22 3D-information-theoretic space for organic molecules with X = halogens or S atoms (hexagons), amino acids (circles), pharmacological molecules (triangles) and other organic molecules including alkanes and ethoxides (squares). Higher to smaller values of CFS are depicted in reddish to blueish colours. Similarly, higher to lower values of CLMC are drawn with bigger to smaller ball sizes (left). Detail of the previous graph for the same molecules except for the set of organic molecules with X or S atoms (right)
The observed similarities in complexity values within specific molecular groups are notable. For example, all organic molecules are represented as bluish squares of approximately the same size; all amino acids are depicted by greenish-yellowish circles, except for those containing sulfur atoms, which exhibit higher LMC complexity values. Similarly, most pharmacological molecules are shown with reddish symbols of a consistent size. However, exceptions such as alkanes and organic molecules containing sulfur or halogens display a broader range of FS complexity, illustrated by a gradient from bluish to greenish, although their LMC values remain relatively constant.
This uniformity in complexity values underscores the chemical coherence within each group despite their diverse molecular structures. It's important to note that while traditional chemical or biological classifications may group these molecules based on standard nomenclatures or the presence of specific atoms, the information-theoretic space reveals deeper physical or chemical attributes that distinguish these groups. For instance, despite all containing similar types of atoms, organic molecules, alkanes, and amino acids, and subsets of pharmacologically active molecules each exhibit distinct behavior and cluster together in the information-theoretic space, highlighting their unique characteristics beyond conventional classifications.
Our research illustrates that the entropy concepts of delocalizability, uniformity, and order, along with their associated dyadic complexity products (LMC and FS), as articulated through the information-theoretic space, effectively distinguish various physical, chemical, and biological attributes across the array of systems analyzed. Notably, these systems adhere to the universally rigorous constraints of the aforementioned complexity measures, validating our findings. The distinct topological characteristics outlined are particularly promising for aggregating a wide spectrum of both simple and complex entities into a coherent map based on information-theoretic principles. We anticipate that these insights will spur further exploration in this realm. Additionally, this topological map could serve as a novel classification and descriptive tool, potentially integrating with established semi-empirical methods like Quantitative Structure-Activity Relationships (QSAR) to discern variations in the pharmacological profiles of biological molecules. Results of the study have been reported in Ref. [96].
The separability problem: atoms in molecules in fuzzy and disjoint domains
In this study, we explore the classical concept of molecular structure, which often depicts molecules as discrete, rigid entities within three-dimensional space, a perspective supported by experimental techniques like X-ray crystallography. Our investigation focuses on the mathematical aspects of dissecting a molecule into its atomic components using various Atoms-In-Molecules (AIM) frameworks, particularly evaluating their relevance and validity through information theory. We analyze several prevalent AIM schemes in chemistry, such as Hirshfeld's stockholder partitioning and the Bader's topological analysis
For the Hirshfeld approach, we utilize the generalized principle of minimum relative entropy based on the Sharma-Mittal two-parameter functional, which circumvents the arbitrary selection of reference atoms typically used in promolecular calculations. In the context of Bader's topological method, we establish that the Fisher information of the defined atomic regions aligns well with the Principle of Extreme Physical Information, thus avoiding issues associated with Schwinger’s principle, which has shown to be less effective.
Uncertainty measures and the generalized principle of minimum relative entropy
In several science fields, informational concepts such as total correlation, information, separability, distinguishability, and various measures of entanglement are unified under the umbrella of uncertainty measures [109]. These measures rely on a probability space, Ω, and a background measure, µ, which can reflect the physical symmetries of a scenario or represent a known probability distribution about a system. This forms the foundation for the principle of minimum relative entropy, which serves as a guideline for revising or updating prior probability distributions. Essentially, this principle seeks the shortest distance between the actual distribution observed in an experiment and the theoretical distribution modeled from the system. For example, if a system is modeled as a collection of statistically independent subsystems, one can investigate various types of correlations by comparing the distributions from the composite system and its subsystems, tailored to the specific property being examined and our understanding of the system. However, quantum states are inherently capable of generating a diverse array of classical probability distributions based on the measurement choices made. Consequently, it's impractical to formulate a universal expression for an uncertainty measure, as it is heavily contingent on the specifics of the systems, measurement techniques, and the properties under investigation. To navigate this complexity, we employ a generalized principle of minimum relative entropy, allowing for flexibility in addressing the intricate nature of these systems.
where φ is a concave function. If the background measure µ represents a probability distribution, for instance σ, we can interpret ρ as being a prior distribution corresponding to our knowledge of the system and σ becomes a posterior density when new information is considered. Let S (ρ,σ) be an uncertainty measure and a three possible generic types of ensemble. The first case might be a classical ensemble described by continuous probabilities distribution pA, pB ... over a n-dimensional space ∆; the second case a classical ensemble described by discrete probability distributions {pi},{pj}..., where i,j,... ranges over some discrete set X; and the third case one could represent a quantum ensemble described by density operators pA, pB,... on some Hilbert space H. Seeking the minimum direct distance between the system under consideration and a model system (e.g., experiment compared against a theoretical model or the whole system against to its constituting parts), we may use an Euler-Lagrange variational principle:
where ρ corresponds to the probability distribution or a density
operator of the system under consideration,
Equation (22) is particularly useful for analyzing Atoms-In-Molecules (AIM) schemes, such as Hirshfeld and Quantum Theory of Atoms in Molecules (QTAIM). These schemes treat the molecule as a composite of N atomic constituents, each represented by a probability distribution in Euclidean space, denoted as {ρA, ρB, …, ρN}. There are significant distinctions between these methods in how the atomic charge densities are treated within the molecular space. In the Hirshfeld scheme, atomic densities overlap within the molecular domain, whereas in Bader’s QTAIM, these densities are distinct and non-overlapping. This difference fundamentally affects the representation of intramolecular interactions and the type of information about these interactions that is retained in the atomic domain after the partitioning procedures.
Stockholder partitioning aim scheme: fuzzy atomic domains
In chemistry, significant interest revolves around understanding the transformation of isolated atoms when they integrate into molecules, manifesting in phenomena like polarization, hybridization, and ionic character. The stockholder partitioning scheme, commonly used in chemical modeling, traditionally does not account for correlations between subsystems, which are critical for chemical reactivity. To bridge this gap, we utilize the concept of general relative entropy, a robust uncertainty measure that effectively captures the essential correlations between a molecule and its constituent parts, such as Hirshfeld atoms. This approach ensures a more accurate reflection of the interactive dynamics within the molecule, crucial for understanding its chemical behavior.
On the other hand, any general relative entropy must obey the semisymmetric property such that:
Therefore, by use of Eq. (23) for the general relative entropy one may obtain the stockholder partition scheme defined by Hirshfeld:
where the weighting function ω was originally obtained by X-ray crystallography techniques. In contrast, we have shown that only information-theoretical properties are necessary.
Next, we apply the generalized principle of minimum relative entropy Eq. (22) by use of any uncertainty measure which fully complies with Eq. (21), for instance the Sharma-Mittal relative entropy [110a], we obtain
Eq. (25) is the Sharma-Mittal
relative entropy of the Stockholder partition AIM scheme. When q → 1 and α → 1
then
Topological AIM scheme: Disjoint atomic domains
The Atoms in Molecules (AIM) scheme, developed by Bader and known as the "Quantum Theory of Atoms in Molecules" (QTAIM), utilizes the topological properties of electron probability distributions in position space, denoted as ρ(r), normalized to unity. This approach divides the Cartesian space into distinct atomic basins without any overlap or mixing, which is why it is referred to as a disjoint AIM scheme. A key feature of this method is the boundary condition of zero flux for the gradient of the electronic charge density, ρ(r), ensuring that each basin is well-defined and separated from others. This unique partitioning allows for a clear delineation of atomic territories within molecules based on electron density contours.
In this methodology, atomic basins ΩA and ΩB are distinctly isolated for a diatomic molecule, providing clearly delineated marginal density distributions for each "atom." This isolation enables the use of information-theoretic properties such as Shannon information entropy to investigate correlations between these subsystems. However, for our current analysis, we opt to focus on Fisher information due to its established connection with generalized relative entropy and compatibility with the Fisher-Rao metric, as indicated by Frieden [73]. Fisher information is notably advantageous for its local sensitivity-it accurately measures density gradients and responds to oscillations in electronic distributions, thereby indicating more localized densities, reduced uncertainty, and improved precision in pinpointing particle localization]. These qualities are crucial for understanding chemical reactivity. Additionally, the Euler-Lagrange equation, which incorporates Fisher entropy as an uncertainty measure and involves second-order derivatives, is typical of many natural mechanistic theories, emphasizing the importance of Fisher Information. This metric, characterized by its shift invariance, plays a vital role in the chemical analysis and offers profound insights into the interatomic interactions and reactivity patterns within molecules.
For a bipartite system we employ Eq.
(27) and the fact that the partition is exhaustive, i.e.,
where Ω A and Ω B are the respective atomic basins in real space and the Fisher entropy of the molecule is the sum of the atomic Fisher entropies. Assuming that the atomic densities ρ i , where i stands for the i-th atom, must be continuously differentiable functions in their respective atomic regions, by use of the Green’s theorem it is straightforward to prove that atomic Fisher measures arise provided that the local zero flux condition is fulfilled:
Next, we can obtain the Euler-Lagrange variational principle, by employing the Fisher information of the molecule as the uncertainty measure:
along with
where ρ is the charge distribution normalized to one and Ω is the spatial
molecular domain. Then, by use of the definition for the functional derivative
which means that the Fisher information of the Bader’s atoms Eq. (32) fulfills all the same properties of the optimal Fisher information measures for the whole system along with the local zero flux condition. The latter should be considered an important result of this work: Fisher information for the molecule and the Fisher information of the Bader’s atoms conforms to a general theory based on the “Principle of Extreme Physical Information” (EPI) [73] by which fundamental equations define atoms in molecules, or any subsystem in bigger systems satisfying Eq. (28), this is in contrast with the Schwinger variational principle used by Bader in his topological definition.
We have explored the application of Information Theory to the task of molecular separability, providing a rigorous theoretical basis for using Atoms in Molecules (AIM) schemes to partition molecules into atoms, referred to as noumenons, i.e. theoretical constructs comprehensible to the intellect but remain beyond the grasp of sensory perception. By utilizing Euler-Lagrange variational equations, we have justified the effectiveness of these schemes in chemical partitioning. Moreover, we've refined the stockholder partitioning approach by eliminating the need for selecting reference promolecular atoms arbitrarily.
Additionally, we have applied Information Theory to the topological partitioning approach, specifically through Fisher information. The analysis reveals that the Fisher information of a whole molecule and that of Bader’s atoms collectively support a robust theoretical framework based on the "Principle of Extreme Physical Information." This principle's foundational equations play a crucial role in defining the atomic structure within molecules, aligning with the theoretical constructs of AIM and providing a consistent and scientifically sound method for analyzing molecular components. Results of the study have been reported in Ref. [97].
Non-local chemical phenomena
Quantum entanglement
Correlation energy as a measure of non-locality: helium-like systems
This study elucidates the intrinsic quantum nature of correlation energy, derived from the wave function independently of any external Hamiltonian. We address the traditionally vague concept of correlation energy by linking it to the quantum notion of nonlocality, identifying it with non-dynamic correlations present in states with a Slater rank greater than one. By associating correlation energy with quantum entanglement measures like the von Neumann and linear entropies, we establish a direct connection between these phenomena.
Particularly, our findings reveal a clear one-to-one correspondence between entanglement and correlation energy in helium-like systems across various atomic numbers (Z). We provide numerical evidence supporting a linear relationship between correlation energy E corr and quantum entanglement for different atoms within the helium isoelectronic series, utilizing configuration-interaction type wave functions that embody high levels of correlation. This alignment underscores the deep interconnection between quantum entanglement and the concept of correlation energy in atomic systems.
The concept: electron correlation
It should be stated that electron correlation in many-electron systems is a fundamental aspect of atomic behavior, intricately linked to quantum entanglement and the concept of non-locality, rather than being a mere artifact of energy computation or a methodological discrepancy arising from comparing variational bounds. In the Hartree-Fock (HF) approach, electrons are treated as interacting only through their average fields, essentially assuming uncorrelated electronic motions. This simplification leads to a discrepancy between the calculated HF energy and the exact non-relativistic energy of the system. This discrepancy is defined as the correlation energy, reflecting the missing energy due to the lack of explicit electron-electron interaction modeling in the HF approximation.
where E0 is exact the true ground-state energy and EHF is the Hartree-Fock energy; its maximum value is zero and corresponds to an independent particle state. It is important to remark that Eq. (33) only represents a practical measure of correlation energy solely accounting for the energy deviation of the independent particle model (HF) from the ones including the so called “electron correlation”, which in turn represents the physical phenomenon of electron interactions.It was Collins who first associated the correlation energy to an informational entropy [112a], which is different to the thermodynamical one. The conjecture by Collins that the correlation energy 𝐸corr is proportional to the information entropy has been amply examined in the literature [112a]. We will discuss below that the independent particle state is equally well characterized by null linear and von Neumann entropies of the one-density matrix, non null quantum entropies (entanglement) occurs when electron correlation is taken into account [112b]
Various methods have been developed beyond the Hartree-Fock (HF) model to address its limitations in accounting for electron correlation. One such method is the Full Configuration Interaction (FCI), which attempts to solve the Schrödinger equation exactly within a specified one-electron basis by employing a linear combination of all possible Slater determinants (LCSD). This approach is highly accurate but computationally feasible only for small systems due to the exponential increase in possible electron configurations as the number of electrons grows.
In FCI and similar LCSD methods, the treatment of electron correlation is purely based on the wave function; the Hamiltonian of the system is not altered. Historically, electron correlation was often seen as a methodological artifact arising from using an inadequate trial wave function, such as a single Slater determinant in the HF approach. This led to a misconception that electron correlation did not correspond to any physical reality.
To better understand electron correlation, it's useful to distinguish between non-dynamic (or static) and dynamic correlation. Non-dynamic correlation accounts for the energy discrepancy when the wave function is represented by only one determinant, reflecting situations where multiple configurations are fundamentally important, such as in bond-breaking processes. Dynamic correlation, on the other hand, deals with the short-range interactions among electrons due to their Coulombic repulsions and is treated in methods that include perturbation from the mean field, such as in coupled-cluster or multi-reference methods.
It's crucial to recognize that in practical computational chemistry, there isn't an explicit division between dynamic and static correlations; both are model aspects of electron correlation captured differently by various computational strategies. Notably, static correlations are explicitly considered in LCSD methodologies, emphasizing the necessity to use these advanced methods for systems where single determinant approaches like HF fail to provide accurate results. In contrast, dynamical correlations are always present when ameliorating the wave function in any post-HF methods or by use of density functional theory (DFT) functionals
Non-locality - quantum entanglement
In recent advancements, a reevaluation of electron correlation through the lens of quantum information theory has led to identifying it as an entanglement phenomenon [112b]. This perspective emphasizes that entanglement, a fundamental quantum property, encapsulates non-dynamical correlation within electronic systems. Our study particularly focuses on helium-like atoms with varying nuclear charges, ranging from 1 to 10, illustrating how entanglement measures such as linear and von Neumann entropies correlate with electron correlation.
Despite utilizing compact one-electron bases to construct full configuration interaction (FCI) wave functions, our findings are sufficiently accurate regarding entanglement and highly correlated concerning energy values. This relationship between correlation energy and entanglement underscores a tangible physical interpretation of non-dynamical correlations, attributing them to the inherent quantum property of entanglement, rather than mere mathematical artifacts of computational chemistry models. This shift in understanding brings a deeper insight into the quantum mechanical behavior of electrons in atoms and molecules, linking traditional chemical concepts with quantum information theory.
Entanglement measures
A pure state of a system constituted by N identical fermions can be described by a single ket vector, |Ψ〉 , so that the associated density matrix is fully defined as ρ=|Ψ〉〈Ψ| . This state is non-entangled, i.e., separable, if it has Slater rank equal to one. Meaning that it can be expressed as a single Slater determinant constructed with N normalized and orthogonal single-particle states {|i〉,i=1,…,N}. Otherwise, the state is called an entangled state.
Several measures quantify the amount of entanglement for a pure state |Ψ〉 of a system with N identical fermions. Linear entanglement measure: Let |Ψ〉 denote a pure state of N identical fermions. The single-particle reduced density matrix, obtained by taking the trace over N − 1 particles, is then: ρr=Tr2,3,…,N(|Ψ〉〈Ψ|
Linear entanglement measure
The linear entropy of ρ r , given by
leads to a practical quantitative measure ξ [ρ r ] for the amount of entanglement of the density matrix state ρ r , namely
which is normalized to the range [0,1]. This quantity vanishes if and only if the state |Ψ〉 has Slater rank 1, and is therefore separable.
According to the above, the linear entanglement measure of the single particle reduced density matrix ρ r of fermionic systems can be expressed in terms of the natural spin orbitals through Löwdin’s spectral decomposition of the ρ r as
where M stands for the dimension of the basis set. It is
worth mentioning that closed-shell atomic systems are commonly represented
through a double occupied density matrix, i.e.
von Neumann entanglement measure
For a pure state of N identical fermions, |Ψ〉, the von Neumann entropy of a single-particle reduced density matrix ρ r , obtained by taking the trace over N −1 particles ρr=Tr2,3,…,N(|Ψ〉〈Ψ|), is given by
This entropy allows us to quantify the amount of entanglement of the N-fermion system’s state |Ψ〉:
This entanglement measure constitutes a non-negative quantity that vanishes if and only if the state |Ψ〉 has Slater rank one and is therefore non-entangled. The term −lnN arises from the fact that even in the separable case, the entropy of the reduced single-particle density matrix does not vanish.
As in the previous case, this quantity can be obtained in terms of the natural occupation numbers:
In our study of the helium isoelectronic series, from H− to Ne+7, we have utilized highly correlated electron densities from the works of López-Rosa et al. [113], derived from configuration interaction (CI) wave functions that systematically vary the nuclear charge, Z. The CI calculations employed a [3s,3p,2d] STO basis set, stratifying the electrons among the K-shell and intershell regions to accurately capture the system's electronic behavior.
Our analysis begins by examining the trend of correlation energy, Ecorr, as the nuclear charge within the helium series increases. Fig. 23 showcases this relationship alongside the entanglement measures, specifically the linear and von Neumann entropies, for each member of the series. The results depict a coherent decrease in both the correlation energy and the entanglement measures as the nuclear charge escalates. This synchronous reduction highlights the intrinsic connection between the quantum mechanical properties of these systems and their underlying electronic structure, underscoring the significant role of nuclear charge in influencing both electron correlation and quantum entanglement within the series.
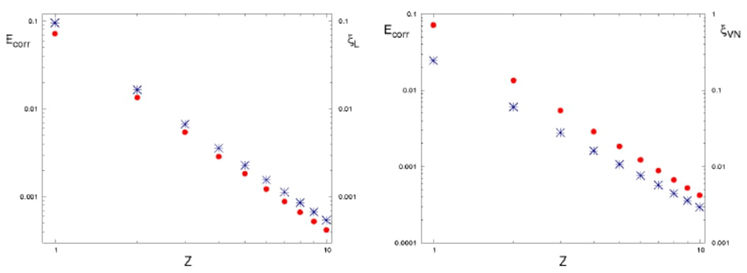
Fig. 23 Correlation energy, Ecorr (dots), and entanglent measures (stars): linear entanglement measure, ξ L , and von Neumann entanglement measure, ξ VN , for helium-like atoms with nuclear charge Z ∈ [1,10].
In order to test numerically that E corr is proportional to ξ L and ξ V N , respectively, we have plotted these values in Fig. 24. We have also obtained least-square fits as shown in Table 2. Interestingly, note from Table 2 that the relationship between entanglement and E corr is close to linearity with correlation coefficients, r, higher than 0.99 in both cases.
Table 2 Parameters and correlation coefficients of Ecorr vs. the entanglement measures.
| a | b | r | |
|---|---|---|---|
| ξ L ξ V N | -2.6203x10-4 4.9278 10-3 | -1.33139 3.39350 | -0.999883 0.99845 |
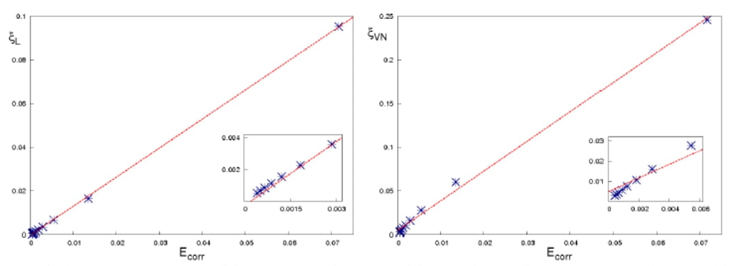
Fig. 24 Correlation energy, E corr , and linear entanglement, ξ L (Z), and von Neumann entanglement ξ VN (Z), for helium-like atoms with nuclear charge Z ∈ [1,10].
We have explored the dynamics between two specific measures of entanglement-linear and von Neumann entropies-and the correlation energy across the helium isoelectronic series, which spans elements with nuclear charges ranging from 1 to 10. Utilizing highly correlated configuration interaction (CI) wave functions, we have observed a consistent trend where both entanglement measures decrease as the nuclear charge increases. This parallel decrease highlights a profound connection: both entanglement and correlation energy fundamentally emanate from the same underlying physical characteristics of atomic systems. Our findings reinforce the concept that in isolated atomic systems, entanglement quantitatively reflects the electron correlation energy, affirming the deep interplay between quantum entanglement and electron interaction dynamics within these systems. Results of the study have been reported in Ref. [114].
The dissociation process of diatomic molecules
We examine into the quantum entanglement characteristics inherent in the dissociation dynamics of selected homo- and heteronuclear diatomic molecules. This analysis is underpinned by meticulous ab initio calculations of the correlated molecular wavefunctions pertinent to the dissociation stages. We meticulously examine the electronic entanglement levels in two critical scenarios: (i) the united-atom limit, where the molecular entities are considered as a single quantum system, and (ii) the asymptotic dissociation limit, where the constituent atoms separate infinitely apart.
Furthermore, we explore how electronic entanglement evolves with the reaction coordinate, providing an in-depth correlation with the various physically significant stages of the dissociation process. Our findings offer new insights into the complex interplay between quantum mechanical properties and traditional chemical phenomena, highlighting the intricate relationship between molecular structure changes and quantum entanglement during chemical reactions.
Entanglement is a ubiquitous phenomenon in composite quantum systems, particularly noticeable when a system dissociates into its components-common scenarios in chemical processes. Yet, the exploration of entanglement properties within chemical systems through quantitative measures remains underexplored. In this study, we focus on the evolution of electronic entanglement during the elementary dissociation processes of both homonuclear (H2, He2, Li2, Cl2) and heteronuclear (HCl) molecules. Employing high-quality molecular wavefunctions obtained through advanced ab initio methods, we utilize the von Neumann entropy of the single-particle electronic density matrix to quantify entanglement.
Our objective is to correlate the changes in electronic entanglement with alterations in other critical physical parameters, notably the total energy, which plays a pivotal role in the dissociation process. The energy’s behavior as a function of the molecular interatomic distance R -the reaction coordinate- provides crucial insights into the dissociation dynamics. By examining the energy against the interatomic distance, two distinct configurations emerge: the united-atom model at minimal distances and the dissociated state at large distances. Our analysis reveals that transitions between these states are reflected not only in energy changes but also in the variations of electronic entanglement, illustrating the profound interconnectedness between molecular structural changes and quantum mechanical properties.
Numerical strategy
This study investigates the dissociation pathways of both homonuclear and heteronuclear diatomic molecules, including H2, He2, Li2, Cl2, and HCl, by employing advanced computational chemistry techniques. The electronic single-particle reduced density matrix, essential for calculating von Neumann entropy and assessing electronic entanglement, was derived using Löwdin's natural orbital-based representation. High-quality correlated molecular wavefunctions, critical for these calculations, were obtained through ab initio methods using the quantum chemical configuration interaction singles and doubles (QCISD) and coupled cluster singles and doubles (CCSD). These sophisticated post-Hartree-Fock methods ensure a variational approach to the computations, contrasting with more common perturbative techniques and guaranteeing proper convergence of energy values.
To effectively model these wavefunctions, we utilized various basis sets including Pople's 6-31G, 6-311G, 6-311++G(d,p) and Dunning's cc-pVTZ and cc-pVQZ. These basis sets are renowned for their ability to accurately represent molecular wavefunctions within the electron density's physically relevant domains. All electronic structure calculations were performed using the Gaussian 03 software suite [62]. The highest level of theoretical accuracy in this study was achieved with the QCISD and CCSD methods using Dunning basis sets, as reflected in the detailed numerical data presented in Tables 3.2.1 and 3.2.2.
With the aim of analysing the behaviors of the von Neumann entropy S(ρr) and the electronic entanglement ξvN during the course of the molecular dissociation, we have tabulated the values adopted by these two quantities, jointly with the values of the molecular and atomic energies, at two limit regimes: at the united-atom representation when R → 0 (Table 3.2.1) and at the asymptotic limit as R → ∞ (Table 3.2.2).
Table 3 von Neumann entropy (Eq. 36) and the entanglement measure (Eq. 37) of the molecular system in the limit R → 0. The entropy of the corresponding ‘united atom’ is also given for comparison.
| Molecular System | United Atom | |||
|---|---|---|---|---|
| Molecule | vN-entropy | ξvN | Atom (statea) | vN-entropy |
| H2 | 0.753 | 0.060 | He (S) | 0.751 |
| He··He | 1.586 | 0.199 | Be (S) | 1.595 |
| Li2 | 1.894 | 0.102 | C (T) | 1.890 |
| Cl2 | 3.565 | 0.038 | Se (D) | 3.554 |
| HCl | 2.940 | 0.049 | Ar (S) | 2.942 |
(a) Notation stands for S-singlet state, T-triplet state, etc.
Table 4 von Neumann entropy (Eq. 36) and the entanglement measure (Eq. 37) and energy of the molecular system in the limit R → ∞. The corresponding quantities associated with the atoms resulting from the dissociation process are also given for comparison.
| Energy | von Neumann entropy | |||||||
|---|---|---|---|---|---|---|---|---|
| System | Molecule | Atoma | Molecule | Atom (S) | Atom (D) | Atom (T) | Tendency | ξvN |
| H2 | -1.000 | -1 | 1.386 | - | 0 | - | 2 ln 2 | 0.693 |
| He··He | -5.805 | -5.805 | 1.445 | 0.751 | - | 0.697 | 1.444b | 0.058 |
| Li2 | -14.865 | -14.865 | 2.024c | - | 1.330 | - | 1.330 (Li)d | 0.231 |
| Cl2 | -919.281 | -919.372 | 3.610c | - | 2.887 | - | 2.887 (Cl)d | 0.083 |
| HCl | -460.117 | -460.186 | 2.996 | 2.953 | - | - | 2.953 (Cl⁻)e | 0.105 |
aSum of the energies of the individual atoms at the dissociation limit (R → ∞).
bEntropy sum of the dissociated atomic states of helium.
cThe molecular entropy value constrained to the double occupation of ρr agrees fairly well with the reported atomic value in the table, see the text.
dEntropy of the dissociated atomic state of highest multiplicity.
eEntropy of the dissociated atomic state for the anion of chlorine.
Hydrogen molecule
In the study of electronic entanglement for the H2 molecule, Fig. 25 illustrates its relationship with the total energy across varying interatomic distances R. Notably, as R exceeds 2 Å, a stage representing molecular dissociation, both the entanglement and energy profiles exhibit similar behaviors. This parallel allows for an insightful examination of the entanglement dynamics, particularly in the united-atom region (R → 0), where the system behaves like a helium-like atom, and in the asymptotic (R → ∞) where the hydrogen atoms lose their Coulombic interactions.
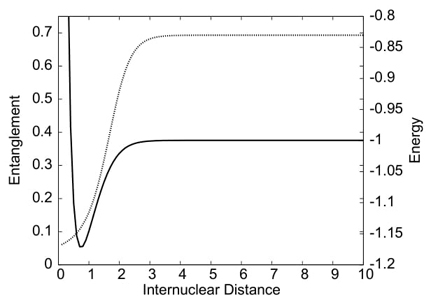
Fig. 25 Entanglement (dotted line) and total energy (solid line) for the dissociation process of the hydrogen molecule.
In the united-atom region, the observed close alignment between the molecular entropy S(ρr) and its counterpart in a helium-like united-atom configuration underlines the high fidelity of the computational results, despite the challenges posed near the R = 0 singularity. As the internuclear distance increases, S(ρr) escalates, stabilizing at a constant value in the dissociation limit, reflecting the persistent, albeit finite, electronic entanglement in the system. Table 3.2.1 provides a quantitative illustration of this phenomenon in the small R limit, while Table 3.2.2 confirms that at large R, the system's energy approximates twice that of an individual hydrogen atom, highlighting the retention of electronic entanglement even at extended separations.
The study of electronic entanglement across the dissociation path of hydrogen molecule reveals distinct behaviors at intermediate, finite values of the interatomic distance (R). In the united-atom limit R → 0, the entanglement within the molecular system diminishes, aligning closely with the entanglement characteristics of a united atom. This reduction is consistent with the ability of the Hartree-Fock approximation to effectively describe atomic eigenstates, indicating minimal entanglement at this stage. Conversely, the behavior of electronic entanglement as R → ∞ - where the molecule dissociates into separate atoms - varies depending on the specific diatomic molecule. This asymptotic behavior necessitates a detailed, molecule-specific analysis to understand the changes in entanglement. Such an investigation underscores the complex relationship between molecular structure, electronic interactions, and quantum entanglement throughout the dissociation process. Results of the study have been reported in Ref. [115].
Chemical reactivity
In this research, we utilize the water molecule and a hydrogenic abstraction reaction to scrutinize quantum entanglement characteristics pertinent to chemistry. The study confirms the necessity of integrating both energetic and quantum-information approaches to thoroughly comprehend the geometry of quantum probability densities in molecular systems and their chemical evolution. Through energy and entanglement hypersurfaces, along with contour maps, distinct phenomena are revealed for these two models. While the energy maps display the well-established stable geometries of the systems, the entanglement maps capture their potential to transition from one state to another. Significantly, the quantum entanglement-chemical reactivity relationship is evident in the entanglement hypersurface of the hydrogenic abstraction reaction, where the transition state denotes a maximally entangled state along the reaction pathway.
Chemical reactivity and entanglement
Traditionally, reactivity in chemistry has been tied to various properties like hardness, activity, electronegativity, and chemical potential, which indicate the extent to which chemical systems can interact. Yet, no single concept definitively encapsulates this phenomenon. Intriguingly, quantum entanglement has emerged as potentially correlated with the general concept of reactivity, reflecting the ability to transition a chemical species from one state to another. Notably, while classical information theory (IT) has helped interpret many chemical phenomena, studies linking quantum IT to chemical reactivity are lacking. Quantum entanglement, a profound manifestation of non-classical correlations between subsystems, suggests that chemical systems inherently possess some level of entanglement, underscoring the importance of exploring its potential connection to chemical stability and reactivity.
This paper aims to (i) determine if total energy and entanglement exhibit similar chemical phenomena, and (ii) investigate the relationship between quantum entanglement and chemical reactivity. For this purpose, we examined the water molecule and the hydrogenic abstraction reaction. These were chosen for their simplicity, involving only three nuclei, which allows for a clear demonstration of the quantum entanglement involved. Additionally, considering water's fundamental role in all living organisms, it is particularly compelling to explore its entanglement-related properties, which have previously received scant attention.
Numerical strategy
This research aims to elucidate two key chemical phenomena using quantum entanglement: (i) the energetic stability of the water molecule, demonstrated through changes in geometric parameters, and (ii) the chemical conversion process in the hydrogenic abstraction reaction. All necessary calculations were conducted at the CISD=full/aug-cc-pvtz level using Gaussian 09 [62].
For the water molecule, we systematically altered the bond angle by increments of 5°, ranging from 35.87° to 175.87°, and varied the bond length between one hydrogen and the oxygen from 0.75 Å to 2.15 Å. The other hydrogen bond was adjusted to obtain the most stable structure while keeping these geometric parameters fixed.
The hydrogenic abstraction reaction, a basic radical abstraction involving atomic hydrogen, was studied by adjusting the bond distance between the incoming (or outgoing) hydrogen and the central hydrogen in increments of 0.15 Å from 0.5 Å to 3.35 Å. This adjustment was made while maintaining the internal angle at 180°. This detailed study of the hypersurface, rather than just the intrinsic reaction coordinate path, aims to deepen our understanding of how entanglement influences chemical processes and vice versa.
Entanglement hypersurfaces of water and the SN1 reaction
Three-dimensional plots were generated to visualize both the energy and the entanglement for these scenarios. Fig. 26 contrasts the energetic profile of the water molecule with its entanglement characteristics, and similarly for the hydrogen abstraction reaction. From these comparisons, we observe distinct behaviors for energy and entanglement. For instance, while energy contours clearly identify the stable geometry of the water molecule (0.96˚A and 104.5◦), the entanglement contours highlight different regions of the hypersurface, which we will discuss in further detail.
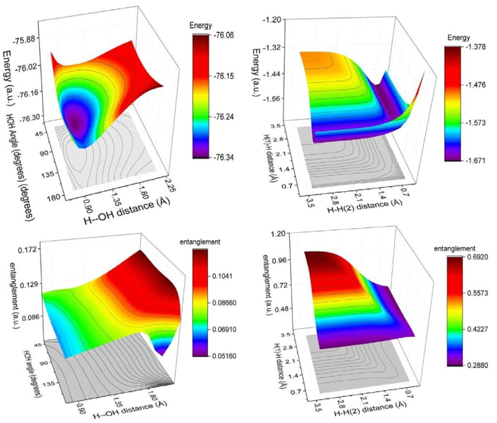
Fig. 26 Hypersurfaces and entanglement values for the water molecule (left) and hydrogenic abstraction reaction (right).
To further explore the quantum entanglement features of the two models, Fig. 27 displays contour maps for both the total energy and the entanglement measure, structured similarly to Fig. 26.
For the water molecule, shown on the left side of Fig. 27, both the top (energy) and bottom (entanglement) contour maps pinpoint the energetically stable geometry, marked with a star symbol. Interestingly, the entanglement map does not correlate directly with energy-based stability; it instead provides insights into the quantum mixedness or purity of the molecule's states.
The hydrogen abstraction reaction, presented on the right side of Fig. 27, illustrates the intrinsic reaction coordinate (IRC) path and the transition state (TS) with a dashed line and a star symbol, respectively. Similar to the water molecule, the entanglement contours for the hydrogen abstraction reaction do not align with the energetic contours. The entanglement measure reveals aspects of the system's state that are distinct from those indicated by its energy profile, highlighting different regions of chemical significance that are not necessarily energetically driven.
This analysis demonstrates that while energy contours are crucial for identifying stable structures and reaction pathways, entanglement contours provide unique insights into the quantum characteristics of molecular systems, emphasizing the importance of considering quantum entanglement in the study of chemical processes.
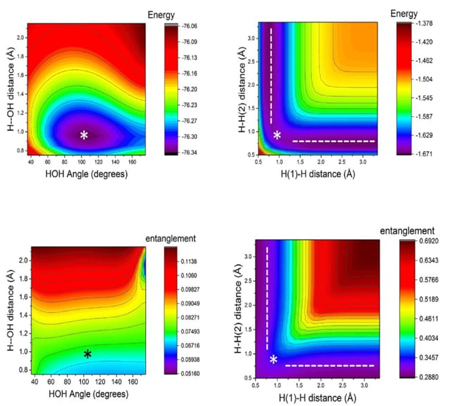
Fig. 27 Contour maps for total energy and entanglement of the water molecule (left) and the hydrogenic abstraction reaction (right).
For the water molecule and the hydrogenic abstraction reaction, focusing on entanglement reveals nuanced behaviors that contrast with conventional energetic analyses:
1. Water Molecule (Fig. 27, bottom-left): The entanglement value generally increases as the H-OH bond length increases, with an exception in a specific area at larger distances and wider angles. Contrary to expectations, at smaller H-OH distances, the entanglement decreases outside the region that defines the energetic stability of the water molecule. This observation suggests that quantum entanglement does not directly reflect the energetic stability but rather the quantum state's complexity and mixedness. A notable point of minimal entanglement occurs at a 180° angle and 1.9Å.
2. Hydrogenic Abstraction Reaction: The contour maps show areas of low entanglement extending beyond the established intrinsic reaction coordinate (IRC) towards shorter H-H distances. Interestingly, within these regions, a maximally entangled transition state (METS), depicted by the star symbol, denotes a state of maximum mixedness. Furthermore, higher entanglement values found beyond the IRC path at greater H-H distances indicate possible alternate reaction pathways. These findings highlight that entanglement can reveal multiple, potentially viable reaction trajectories, each associated with different degrees of quantum mixedness and possibly distinct reaction mechanisms.
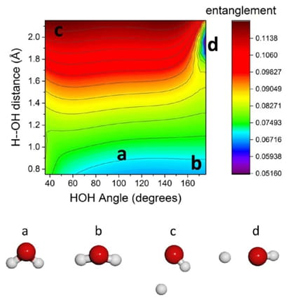
In Figures 28 and 29, the entanglement contour maps for the water molecule and the hydrogenic abstraction reaction are presented alongside specific molecular geometries to illustrate the electronic entanglement response to nuclear rearrangements.
Fig. 28 reveals that the molecular geometry associated with the lowest energy for the water molecule does not coincide with any critical entanglement value (structure a). This disconnection underscores the complexity of quantum behaviors versus classical energetic expectations. For structures b and c, which are associated with higher energy levels indicative of excited states, there is a distinct contrast in their entanglement values. Structure b exhibits lower entanglement compared to structure a, suggesting simpler electronic interactions. Conversely, structure c shows increased entanglement, pointing to more complex electronic behaviors. The minimal entanglement value is observed in structure d, which aligns with the significant geometrical transition at a bond distance of 1.8 to 2.0 Å and an angle of 180°, a configuration indicative of the bond cleavage process. These observations highlight the nuanced relationship between molecular geometry, energy levels, and quantum entanglement. The entanglement contour maps serve as critical tools for identifying regions within molecular systems where quantum effects dominate, influencing both the stability and reactivity of the system. The analysis confirms that while higher energy often correlates with more complex electronic states as indicated by increased entanglement, the specifics of molecular interactions and their rearrangements can lead to unexpected variations in quantum behavior, crucial for understanding chemical reactivity at a fundamental level.
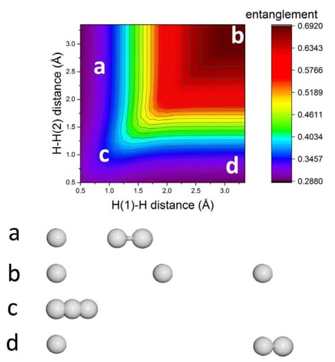
The entanglement hypersurface for the hydrogen abstraction reaction, as depicted in Fig. 29, illustrates unique quantum characteristics that differ significantly from classical energy considerations. In the analysis:
- Structure a, which represents the minimum-energy configuration, surprisingly does not correlate with any critical value for entanglement, underscoring the complex interplay between quantum properties and molecular energetics.
- Structure b, is characterized by being the most-separated molecular geometry, interestingly corresponds to the maximally entangled state. This highlights a key aspect of quantum mechanics where entanglement can be significant even at large molecular separations.
- Structure d, is associated with the lowest entanglement state, representing a scenario of long-distance interaction between two subsystems. This configuration emphasizes that entanglement diminishes as the interaction between subsystems becomes less direct.
- Structure c, is chemically significant as it represents the transition state (TS) of the reaction. Notably, it corresponds to the maximally entangled state along the intrinsic reaction coordinate (IRC) path, further establishing the profound link between quantum entanglement and key chemical transition states.
These observations indicate that while entanglement remains constant beyond certain interatomic distances, it profoundly influences the chemical dynamics at critical points, particularly at the TS. Such insights reveal the fundamental quantum nature of chemical reactions, where entanglement not only elucidates the structure and behavior of molecular systems but also enhances our understanding of reaction dynamics beyond traditional energy-based models.
The connection between quantum entanglement and chemical reactivity provides a profound insight into the molecular behavior and transformation dynamics. Entanglement, particularly in its maximal form, correlates strongly with the reactivity of chemical systems. Highly entangled states indicate a system's greater ability to undergo transitions between different molecular configurations or react with various chemical species, reflecting a high degree of chemical reactivity. Key observations include:
1. Transition State (TS) Reactivity: The transition states, as critical points along the reaction path, exhibit maximal entanglement, which is a quantum signature of their high reactivity. These states, crucial for chemical transformations, are shown to be maximally mixed, as highlighted in reference to Esquivel et al., where the TS represents a peak in entanglement along the intrinsic reaction coordinate (IRC) path.
2. Saddle Points and Higher Excited States: Fig. 16 demonstrates that apart from the TS, there are multiple paths within a chemical reaction that exhibit very high entanglement at what are termed higher-order saddle points. These states correspond to reactions involving highly excited molecular states, suggesting that these pathways, while perhaps less likely under normal conditions, could play significant roles under specific conditions enhancing reactivity.
3. Low Entanglement and Stability: Conversely, lower entanglement values are indicative of less reactive species. For example, the reactants and products in the hydrogen abstraction reaction, represented by structure a in Fig. 29, show minimal entanglement, aligning with their relatively stable nature under normal conditions. Similarly, the aqueous chemical species represented by structure d in Fig. 28, exhibiting lower entanglement, suggests a less reactive or more stable state within the reaction pathway. Results of the study have been reported in Ref. [116].
Concluding remarks
In recent years, there has been a growing interest in analyzing the electronic structure of atoms and molecules using Information Theory. Our ongoing research across various chemical domains has yielded intriguing insights into chemical phenomena through the application of Classical and Information Theory (CIT and QKN). Notably, information-theoretic measures have proven effective in providing simple, yet profound, pictorial descriptions of systems and their processes by highlighting essential behaviors such as the "localized/delocalized" features of the electron density through Shannon entropy, or the "narrowness/disorder" of the distribution unveiled by Fisher-information, or else the "uniformity/deformation" features grasped by the Disequilibrium measure. Additionally, the combined dyadic products of information-theoretic functionals elucidate several aspects of statistical complexity, in both position and momentum spaces. For example, our studies have demonstrated how the IT functionals (S, I and D) can explain the synchronous/asynchronous behavior in the mechanistic progression of elementary reactions or their ability to detect all concurrent phenomena at the vicinity of the state of transient of chemical reactions.
Perhaps one of the most relevant contributions of our work has been to establish rigorous mathematical information-theoretic justifications for employing some of the most prevalent AIM schemes in the field of Chemistry, including Hirshfeld (stockholder partitioning), Bader’s (topological dissection), and the quantum information approach in Hilbert’s space.
The subtle understanding that entanglement not only enriches the classical perspective of chemical reactivity but also introduces a new dimension to predict and manipulate reaction pathways based on quantum mechanical properties is perhaps one of the main achievements of this work. This could potentially lead to novel approaches in chemical synthesis and reaction dynamics, leveraging the intrinsic quantum nature of molecules for advanced applications. These insights demonstrate the crucial role of quantum entanglement in understanding chemical reactivity and mechanism, providing a more complete picture that complements the traditional energy-based descriptions. The unique regions of high and low entanglement across different geometrical configurations suggest complex interplays of quantum states that influence the reaction pathways and stability of molecular systems.
Local and non-local aspects of chemical phenomena have demonstrated through our investigations in several areas of chemistry and related fields that there are strong reasons to believe information science, both classical and quantum, forms a new scientific language to explain essential aspects of chemical phenomena. These new insights are completely inaccessible through any standard methodology, revealing intricate mechanisms in which chemical (and presumably biological) phenomena occur. This opens a promising new area of research as a standalone and robust discipline. Therefore, our main goal is to cultivate fertile ground in the coming years to demonstrate to the physical and chemical communities the existence and benefits of a new language based on the axiomatic interplay between our chemical intuition and theoretical information, which is shaping the science of Quantum Information Chemistry.

