











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Multicomponent reactions (MCRs) are defined as highly convergent one-pot processes in which three or more reagents are simultaneously, sequentially, or consecutively combined to assemble complex products that contain most of atoms coming from reactants (efficient atom economy) [1]. MCRs can be classified into two main groups: a) MCRs based on the use of isocyanides, and b) non-isocyanide-based MCRs [2]. We recently reported a review-type manuscript discussing reactions based on the use of isocyanides, particularly the Ugi three-component reaction and its variants [3]. In 2009, M. Syamala reported a review focused on three-component reactions (3CRs) [4].
Polyheterocyclic structures are found in a wide variety of products with interesting applications in medicinal chemistry, materials science, agrochemistry, optics, etc. [5-8]. MCRs have been considered as synthetic methodologies of choice to construct polyheterocyclic structures which contain heteroatoms mainly like oxygen, nitrogen, and/or sulfur, just to name a few [9].
This review is focused on non-isocyanide-based 3CRs such as Strecker, Biginelli, Mannich, and Petasis reactions, to name the most known, highlighting applications of synthesized compounds, mainly polyheterocycles. It is important to highlight that the present review includes the literature since 2010.
Strecker-type 3CRs
Strecker reaction was performed for the first time by Adolph Strecker in 1850. This 3CR allows the synthesis of α-aminonitriles 5 (Scheme 1) as result of condensation between a carbonylic compound 1 and an amine 2 (e.g., primary or secondary amines, or ammonia) to afford aminoalcohols 3 which after a dehydration performed iminium ions 4, followed by the addition of a cyanide source (HCN, TMSCN, etc.) in the same reactor to give the compounds 5. Moreover, it is known an asymmetrical version of this coupling consisting of sequential combinations of chiral imines, carbonylic components, and cyanide anions but using organocatalysts or chiral metal-based compounds as catalysts to achieve enantio-enriched mixtures of α-aminonitriles. The α-aminonitriles are adequate precursors of α-amino acids. This 3CR owing to its simplicity and atomic economy is appropriate for further transformations [10].
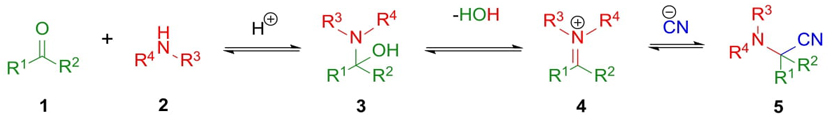
In 2011, Stocker et al. reported the synthesis of various aminoiminohexitols or azasugars (Scheme 2). Some of these compounds show glycosidase inhibitory activity. The synthesis starts from D-ribose 6. The key step of this methodology involved a diastereoselective Strecker reaction between the D-ribose-derived aldehyde 7, NH4OAc, and TMSCN to afford the α-aminonitrile 8 in excellent yield (92%). The major diastereomer was placed in syn configuration (syn/anti 8:1). Diastereomer 8 was isolated and then treated with NaHCO3 and I2 promoting carbamate annulation to afford the bicyclic compound 9. After hydrogenation, and deprotection it was achieved the compound 10. Finally, a hydrolysis of compound 10 completes the total synthesis of the aminoiminohexitol 11 in 36% overall yield [11]. It is worth noting the high stereoselectivity of the Strecker reaction as well as the absence of metal catalysts.
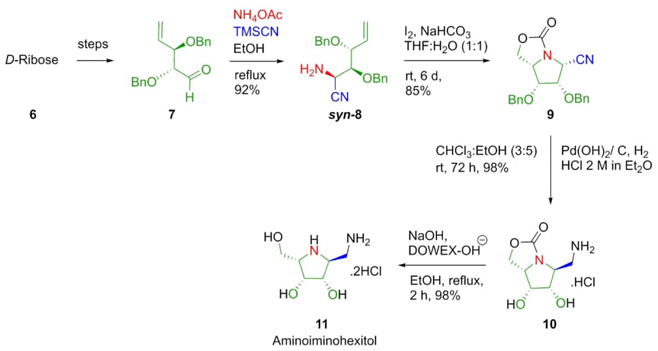
A tandem Strecker-Lewis base-catalyzed reaction was employed by Liao et al. in the synthesis of γ-lactams [12]. As the first step, amino nitrile intermediates 14 were achieved through a sequential combination of aldehydes 12, amines 13, and TMSCN under basic conditions generated in situ (Scheme 3). Then, functionalization of amino nitriles 14 via allylic alkylation was furnished by reacting with methyl acrylate 15 to provide the precursors 16. Thus, the new γ-lactams 17 were synthesized in moderate to good yields (52-92 %) from the intermediates 16 through the assistance of DBU in acetonitrile as solvent. This Strecker reaction was an excellent option for synthesizing highly functionalized aminonitriles that for the allylic-alkylation-cyclization strategy is regioselectively driven by polar solvents such as acetonitrile.
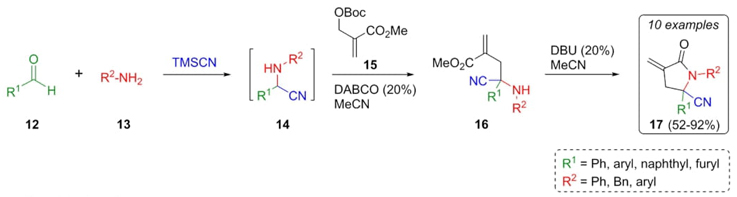
Another nice example of a Strecker reaction is found in a report by Cardona et al. [13]. Aldehyde 19 was previously prepared starting from D-mannose (18) in four steps (Scheme 4). Then, a catalytic hydrogenation of ketal 19 was performed to give the aldehyde 20, which then was subjected to a Strecker reaction in mild conditions using benzylamine and trimethylsilyl cyanide. Finally, nitrile 21 was acetylated under basic conditions affording the piperidine-containing acetate 22. Noteworthy, Strecker reaction was the step key as stereo and regio-selective due to the following reasons:1) it was taken the stereochemistry of compound 20 to carry out the Strecker reaction, where the formation of the iminium ion favored a chelated intermediate in which the cyanide anion performed the addition on the Si face to the double bond forming the diastereomer S as the major product. In the same way, reductive amination at the C-1 position was promoted by the C-5 position of aldehyde.
Khan et al. reported a short and original asymmetric Strecker reaction to synthesize (S)‑Clopidogrel (antiplatelet agent) [14]. 2-Chlorobenzaldehyde (23) in the presence of bis-heterocycle 24 as the amine generated in situ an iminium salt, which was trapped by TMSCN. The chiral alkaloid 25 as an organocatalyst was included. The reaction gave a good yield (92 %) and enantioselectivity (er 78:22). A further Pinner reaction with nitrile 26 achieved compound 27, and its acidic hydrolysis afforded the (S)-Clopidogrel (28) (Scheme 5). Authors suggested that the use of NaF as a polarizer of the Si-CN bond facilitated the nucleophilic attack by CN. Coupled with it, it is noted that the use of this kind of organocatalysts made it possible to perform Strecker reactions in enantioselective manner. This was the first time where a Cinchona alkaloid derivative was used in an asymmetric Strecker reaction, in this case occurred a process of dynamic kinetic resolution to generate quantitative yields in one of the enantiomers [15].
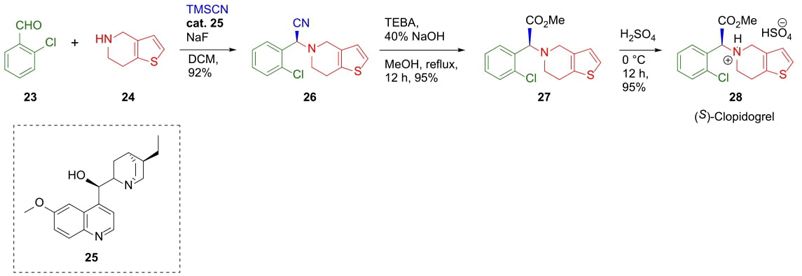
In 2013, Liu, Hao et al. designed an unusual strategy with extreme atom economy towards the synthesis of Trigonoliimine A (hexacyclic bisindole alkaloid isolated from Trigonostemon lii), in which one of the key steps was a Strecker-type reaction [16]. Oxidation of tryptamine 29 gave the ketone 30. Thus, the TMSOTf-catalyzed sequential combination of N-phthaloyl-derivative aldehyde 30, tryptamine (31), and TMSCN gave the Strecker-intermediate 32. Seven-membered ketone 33 was obtained from nitrile compound 32 via Houben-Hoesch-type cyclization in 60% yield. Trigonoliimine A (34) was provided in 85% yield by treatment of seven-membered ketone 33 with hydrazine in ethanol (Scheme 6). It is noteworthy that Strecker reactions with ketones are hard to perform.
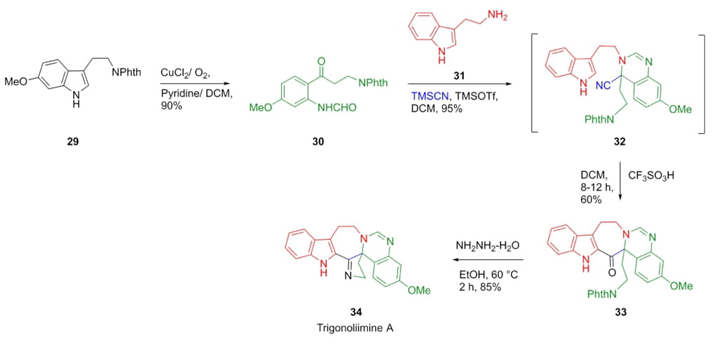
In 2015, Grygorenko et al. continued developing their strategic route called tandem Strecker Reaction-Intramolecular Nucleophilic Cyclization (STRINC) (Scheme 7) [17]. Thus, pyrrolidine-derived γ-bromoketone 39 was synthesized from L-4-hydroxyproline 38 after several chemical steps. After optimization, STRINC process was carried out between ketone 39 and reagent 37, previously generated through a combination between amine 35 and nitrile 36, affording bicyclic aminonitrile 40. Acid hydrolysis of aminonitrile 40 and deprotection of acid 41 completed the synthesis of bicyclic amino acid 42 in an overall yield of 7 % after 10 steps. This one is a peculiar kind of methodology giving good atom economy to develop bicyclic scaffolds.
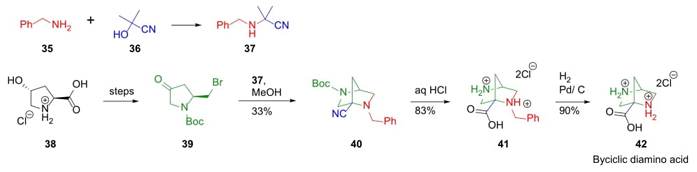
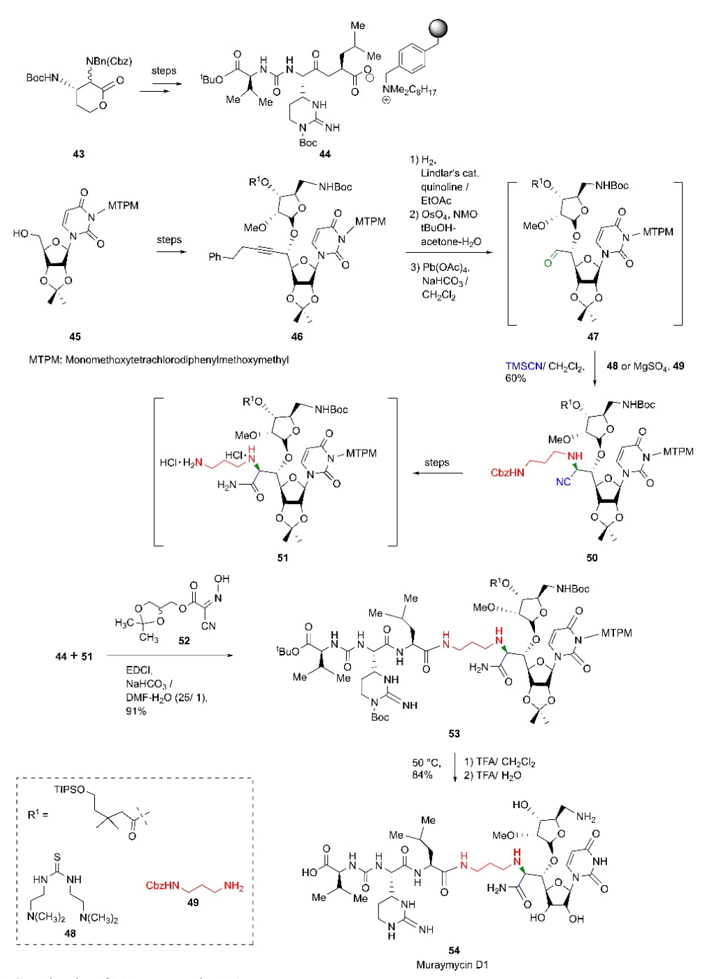
In 2016, Kurosu et al. reported a stereocontrolled total synthesis of Muraymycin D1 [18]. Due to the complexity of the latter, its synthesis was divided in two crucial sides. a) Starting from carbamate 43, it followed a sequence of reactions such as ureido-muraymycidine tripeptide builded by epimerization-lactone opening and a cyclic guanylation as key steps. b) The other moiety, starting from carbohydrate 45, the chemical steps comprise a complex of 3-aminopropyl amino acid involving several processes such as oxidation, Carreira's asymmetric alkynation, β-selective ribosylation, hydration with HgCl2, a selective deprotection, and a stereoselective Strecker reaction as one of determining steps of this route. The reaction between aldehyde 47 (precursor of Strecker reaction) with Cbz-monoprotected 1,3-diaminopropane 49 and TMSCN catalyzed by thiourea 48 or magnesium sulfate produced the desired aminonitrile 50 with absolute selectivity in (S)-diastereomer form (Scheme 8). Authors proposed thiourea 48 or magnesium sulfate to promote separation of the mixture of enantiomers. The join of the peptidic chains 44 and 51 was promoted by glyceroacetonide-oxyme 52 through a decomplexation process with compound 44. Finally, after a deprotection step of compound 53 afforded Muraymycin D1 (54). It is worth highlighting the complexity of this synthetic strategy, its stereoselectivity, and the implementation ‘for the first time’ of a stereoselective Strecker reaction as the key step. Also, it played an extremely important role in the joining of fragments 44 and 51, leading to a high proportion of the (S)-diastereomer.
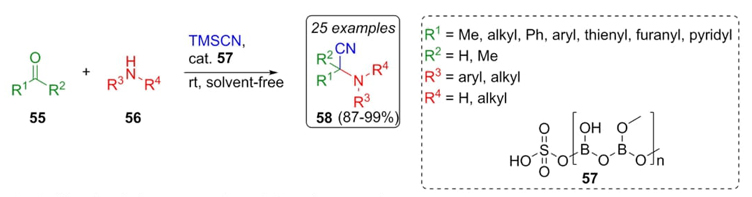
Chaturbhuj et al. reported an efficient and low toxic strategy involving the use of a solvent-free organocatalyzed Strecker reaction between aldehydes 55, amines 56, TMSCN and borate catalyst 57 at room temperature to afford in up to 30 minutes the products 58 shown in Scheme 9 in good to excellent yields (87-99%) [19]. In scope, different electron-donating and electron-withdrawing groups were evaluated, as well as polar and non-polar solvents, finding that the best reaction condition was under a solvent-free environment. In addition, the effect of the catalyst on the reaction was proven using a panel of Lewis and Brønsted acids, resulting to be the sulfated polyborate the best one. Finally, many advantages of the trimethylsilyl cyanide (TMSCN) as a cyanide source were demonstrated, but mainly safe use, ease of handling, and effectiveness.
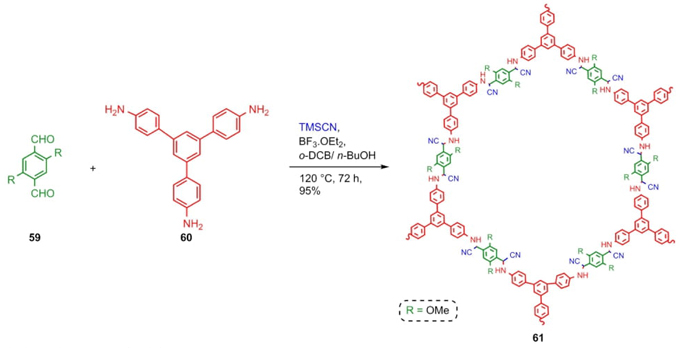
Covalent Organic Frameworks (COFs) belong to a special type of crystalline porous materials, that have caught the attention of synthetic and material chemists for their applications such as catalysts or gas storage materials [20]. The approach of Chen, Dong et al. involved a mixture of aromatic aldehydes 59, amine 60, and TMSCN catalyzed by a Lewis acid to afford the products 61 in 95 % yield (Scheme 10) [21]. There are currently a few examples of COFs designed and synthesized through multicomponent reactions due to their complexity, but these methods turn out to be good choices for the preparation of these types of materials.
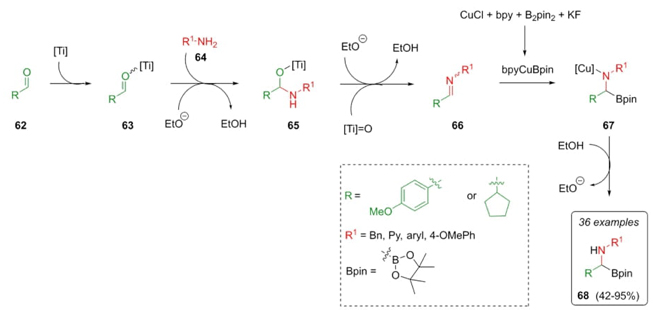
Marder et al. reported a chemoselective strategy to synthesize α-amino boronates via a new named Borono-Strecker reaction [22]. Thus, aldehydes 62 coordinate with titanium to give intermediates 63, which were combined with amines 64 in the presence of Lewis acid Ti(OEt)4 promoting imine’s 66 formation after an ethanol release in compounds 65. A proton source is necessary for the hydroboration of imines 66. This latter is due to the formation of a fluoride assisted by ethoxide anion, activating B2pin2 (bis(pinacolato)diboron) for copper transmetalation in compound 67, using MTBE as a solvent to give the α-aminoboronates 68 in moderate to good yields (42-95 %) (Scheme 11). As seen, the key idea was to use a Lewis acid together with a proton source (Brønsted acid) to avoid side reactions between carbonylic compounds and B2pin2.
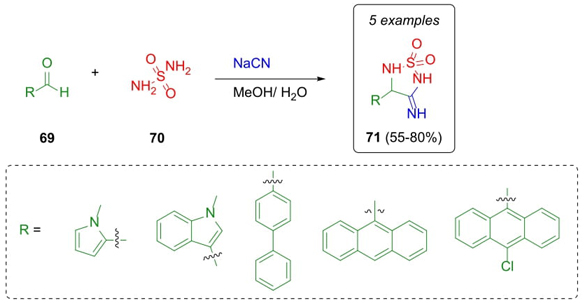
In 2022, Başkan et al. reported the synthesis and in vitro evaluation of 3-imino-sulfahydantoins on bacteria and cytotoxicity studies [23]. These compounds were prepared through a Strecker reaction by a combination of different aromatic and heterocyclic aldehydes 69, sulfamide 70, and sodium cyanide to give 1,1-dioxides 71 in moderate to good yields (55-80 %) (Scheme 12). These compounds exhibited potential antibacterial properties and cytotoxic effects on SPC212 cells related to malignant pleural mesothelioma, a type of lung cancer.
Biginelli-type 3CRs
In 1893, Pietro Biginelli made possible another kind of a three-component reaction by protonation of carbonyl compounds 72. Then, protonated aldehydes 73 were combined with urea or thiourea (74) to give aminoalcohols 75, which after dehydration performed iminium ions 76. The addition of a β-ketoesters 77 (e.g. ethyl acetoacetate) achieves 3,4-dihydropyrimidin-2(1H)-ones 80 after a cyclization step of compounds 78 and dehydration of intermediates 79 (Scheme 13) [24]. Also, this kind of condensation could be catalyzed by many Brønsted or Lewis acids. Asymmetric synthesis of many dihydropyrimidinones has been also reported to develop new compounds with pharmaceutical interest [25].
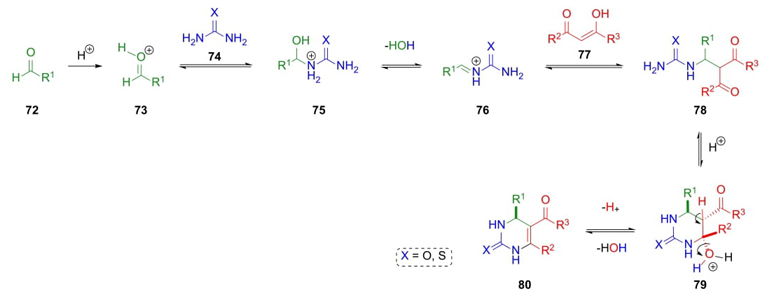
In 2010, Tellitu, Dominguez et al. developed an interesting methodology to achieve furo[3,4-d]pyrimidinones 88 using a Biginelli reaction as the key step [26]. After corresponding optimizations, 5-carboxamido-dihydropyrimidines 84 were afforded from 3-ketoamides 81, aldehydes 82, and urea-derivates 83 using chloroacetic acid as catalyst, under solvent-free conditions (Scheme 14). Compounds 84 in the presence of PIFA [phenyliodine(III) bis(trifluoroacetate)] promote formation of iodine complexes 85. Then, a 1,5-hydride shift achieved intermediates 86, where the allylic position was attacked by oxygen of amide fragment to give the iminolactones 87. Finally, a hydrolysis step developed corresponding furo[3,4-d]pyrimidine-2,5-diones 88 in moderate yields (17-65%). PIFA plays an important role in intramolecular oxycarbonylation. Also, electronic or steric effects into aryl groups promote the corresponding yields.
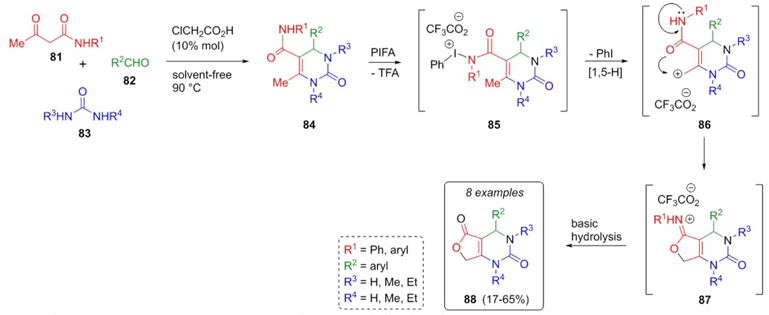
A closer work was reported by Singh et al. in 2010, where it was performed for the first time a silica-sulfuric acid-catalyzed Biginelli-type reaction [27]. It was proposed the use of β-oxodithioesters as β-dicarbonyl components. After corresponding optimizations, oxodithioesters 91, aldehydes 92, and urea (93) were combined with SiO2-H2SO4 as acid catalyst to generate the Biginelli-derivatives 97 in moderate to good yields (65-85%). It was proposed a plausible mechanism where condensation between aldehydes 92 and urea (93) performed iminium cations 95. After the addition of dithioester enols 91 (previously afforded through condensation between ketones 89 and thioate 90), an intramolecular cyclocondensation, and dehydration performed dihydropyrimidinones 97 (Scheme 15). It has been used different electron-withdrawing and electron-releasing substituents. The authors mentioned that it was unable to know the role of thiourea in thiocoumarin-derivatives. However, it is worth noting the role of silica-sulfuric acid to promote the involved cyclizations in this methodology.
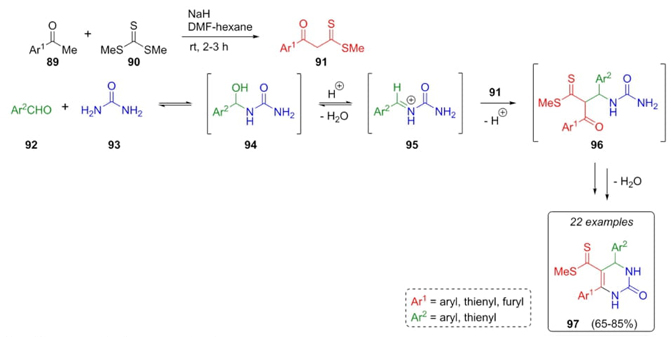
In 2010, Ji et al. reported the synthesis of a series of novel dihydropyrimidinones through a Brønsted-base (t-BuOK)-catalyzed Biginelli-type reaction [28]. In the first pathway, aldehydes 98 were combined with ketones 99 to give the compounds 100 under basic conditions. Then, the addition of thiourea (101) achieved intermediates 102, which after a ring closure afforded the compounds 103. Finally, a dehydration step performed the thiones 104 (Scheme 16a). In the second pathway, a condensation of aldehydes 98 and urea (101) gave the compounds 106. After the addition of ketones 107, cyclization of compounds 108, and dehydration of cyclic compounds 109, dihydropyrimidin-2(1H)-ones 110 were obtained (Scheme 16b). After several optimizations, two series of compounds were prepared depending on the reagents used. In the same way, the mechanism may take two different pathways in function of the reagents used (Scheme 16 a) thiourea, or b) urea), where the important intermediates are furnished after corresponding condensations in each pathway. It is important to note that this was the first Biginelli reaction developed under basic conditions.
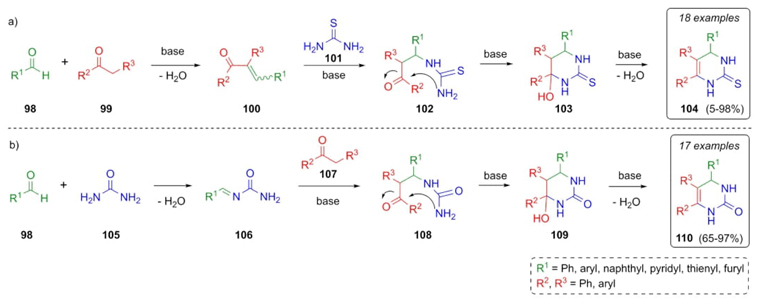
Scheme 16 Brønsted base-catalyzed synthesis of a) thiones, and b) 4,5,6-triaryl-3,4-dihydropyrimidin-2(1H)-ones.
The search behind better organocatalysts with associated lower costs and toxicity, but even with enantioselective efficiency led to Moorthy and Saha developed a Biginelli reaction including mechanistic studies of sterically hindered organocatalysts (Scheme 17) [29]. After exhausting studies, it was found that catalyst 112 worked in high selectivity on Biginelli reaction from a series of electron-rich and electron-deficient aldehydes combined with ethyl acetoacetate (111) and urea to afford the products 117 in up to 66 % yields but in excellent enantiomeric excess (94-99 %). Mechanism pathway proposed involves condensation between catalyst 112 and dicarbonilic compound 111 to give the enamine 113. Then, nucleophilic attack from enamine 113 to iminoamides 114 performed pyrrolidinium cations 115. Finally, cyclization of precursors 116 achieved the products 117. Despite the moderate yields, the enantioselectivity found was good because the catalyst used is bulky and contains a proton donor group, which favors the re-facing attack of complex 113 towards the iminoamide 114.
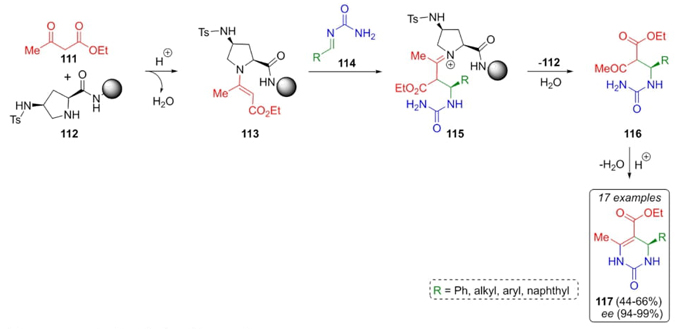
Gogoi, Baruah et al. developed an ultrasound-assisted Biginelli-type reaction [30] to efficiently synthesize some new 3,4-dihydropyrimidin-2(1H)-ones and 3,4-dihydropyrimidine-2(1H)-thiones. In this one-pot reaction, steroidal ketone 118 was combined sequentially with aldehydes 119, urea, or thiourea (121) using ultrasound conditions and sodium ethoxide as the base. Biginelli-species 123 were formed with excellent yields (85-94 %). It was proposed a plausible mechanism that proceeds through an aldol condensation between ketone 118 and aldehydes 119 to give the compounds 120, which through a nucleophilic attack by urea or thiourea (121) gave intermediates 122, which after releasing water furnished the steroids 123 (Scheme 18). It is well-known that ultrasound-assisted reactions have an important impact on green chemistry, giving added value to one-pot reactions. In the particular case of this work, ultrasound energy improved the yields probably due to mechanical stress in the process.
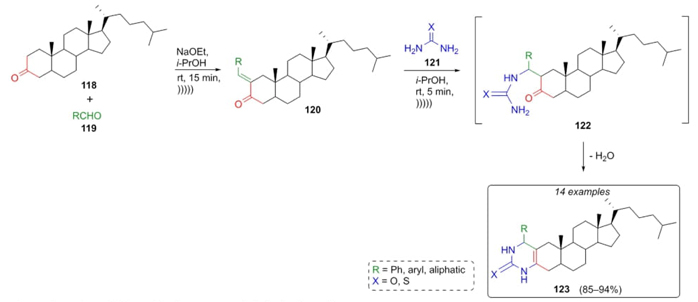
The synthesis of a dozen of new Biginelli compounds developed by Breit et al. involved the use of alkenes to generate aldehydes through a hydroformylation process catalyzed by a rhodium-xantphos complex [31]. After corresponding optimizations, alkenes 124 were combined with ethyl acetoacetate (126) and urea (125), HCl was added in catalytic amounts (Scheme 19). Functionalized dihydropyrimidones 127 were synthesized in moderate yields (52-78 %). Rh-xantphos catalyst usually used in hydroformylation reactions was employed here in a Biginelli reaction and by-products derived from carbonylation reaction were not observed, except when large chain alkenes 124 were used, resulting in low yields for the Biginelli products.
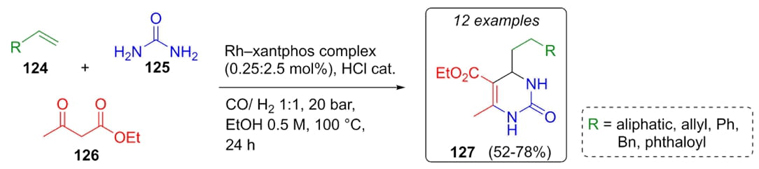
Another example of an eco-friendly methodology is the synthesis of Biginelli products under ball-milling assistance reported by Mal et al. in 2015 [32]. First, the aldehydes 129 were prepared through alcohols 128 oxidation under solvent-free conditions by ball-milling using a catalytic amount of oxone (peroxymonosulfate ion), KBr, and TEMPO [(2KHSO5·KHSO4·K2SO4)-2,2,6,6-tetramethylpiperidin-1-yl-oxy radical]. With aldehydes 129, and reagents 130 and 131 in hands, Biginelli reaction proceeded also under solvent-free conditions and using a ball-milling device to give products 132 in moderate to good yields (78-95 %) (Scheme 20). Acid subproduct in the oxidation reaction served as the catalyst in Biginelli reaction. Notably, this method employed electron-withdrawing and electron-releasing aldehydes to afford new dihydropyrimidones in a one-pot process. Also, regioselectivity was directed by using N-methyl urea to obtain just a single regioisomer.
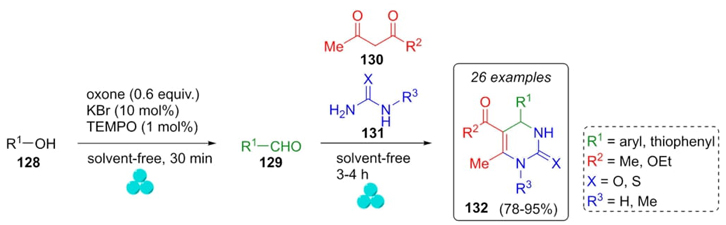
Silvani et al. developed an organocatalytic asymmetric method for the construction of spiro-compounds via a Brønsted acid-catalyzed Biginelli-type reaction [33]. Thus, isatin derivatives 133 were combined in a one-pot process with β-ketoesters 134 and urea (135) using the chiral organocatalyst 136 to give the spiro(indoline-pyrimidine)-diones 137 in moderate to good yields (49-93%) with moderate ee values (50-80%) (Scheme 21). This report was the first one to describe an asymmetric Biginelli-type reaction from ketones under acid-organocatalytic conditions in which 3,3’-positions of the catalyst have a strong influence on the yields and ee. Best yields and enantioselectivity were observed for the N-methyl isatin derivative rather than others with substituents like benzyl.
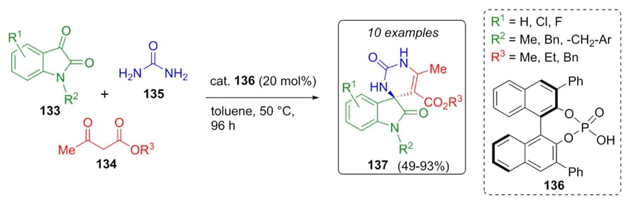
Scheme 21 Synthesis of spiro(indoline-pyrimidine)-diones via an organocatalytic asymmetric Biginelli reaction.
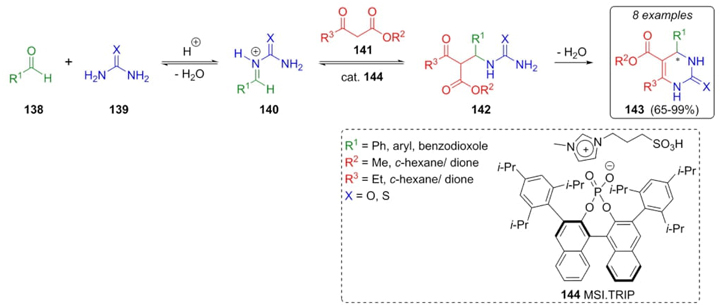
Amarante, Neto et al. developed a synthetic strategy where a chiral catalyst was used in a Biginelli reaction [34]. The catalyst 144 containing a chiral phosphoric acid moiety in ionic liquid media resulted in a creative method called Asymmetric Counteranion-Directed Catalysis (ACDC). Thus aldehydes 138 were combined with urea or thiourea (139) to give the intermediates 140. Further addition of keto-esters 141 to compounds 140, and dehydratation of precursors 142 gave good to excellent yields (65-99 %) and ee (37-99 %) of compounds 143. It was proposed that Biginelli reaction followed the iminium pathway (Scheme 22). The catalyst 144 was recovered in 95 % amount and reused many times. This feature joined to solventless conditions results in another example of an environmentally friendly multicomponent reaction toward heterocyclic compounds.
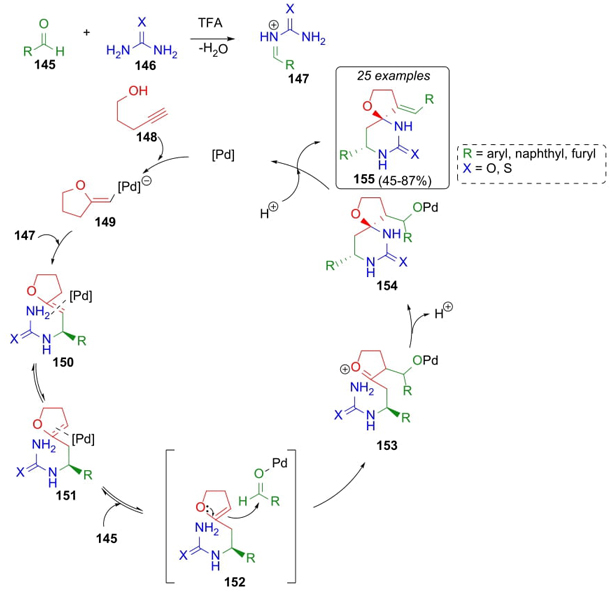
Yu, Ying et al. used a cyclic enol ether as the main reagent in a Biginelli reaction [35]. In this approach, it was necessary to optimize reaction conditions, being found as the best catalyst PdCl2 and trifluoroacetic acid as solvent. Sequential combination of aldehydes 145 with alkynol 148 and urea or thiourea (146) afforded the spiro-compounds 155 in moderate to good yields (45-87 %). A plausible mechanism involves a catalytic cycle involving the use of palladium (Scheme 23). As the first step, vinyl palladium complex 149 was trapped by N-acyliminium intermediates 147 through an intermolecular addition furnishing intermediates 150. Then, palladium catalyst promoted the formation of endocyclic enol ethers 151 via double bond isomerization. Enol ethers 151 perform a nucleophilic attack to aldehydes 145 to give the intermediates 152. After intramolecular cyclization of complexes 153, protonation, and dehydration of intermediates 154, the catalytic cycle was completed regenerating the palladium complex 149. Formation of spiro compounds was achieved in a combination of metal and Brønsted acid catalytic cycle in excellent enantioselectivity and with the best yields in a wide variety of electron-donating and withdrawing groups.
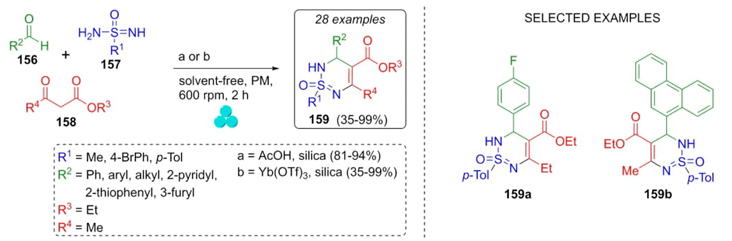
In 2021, Bolm et al. reported the synthesis of a series of dihydro-thiadiazine-oxides via a one-pot mechanochemical Biginelli-like process [36]. Optimal reaction conditions were found and were using acetic acid and silica gel as Brønsted and Lewis acids, respectively (Scheme 24). Thus, towards series a, aldehydes 156, sulfonimidamides 157, and ethyl acetoacetate (158) were placed in acetic acid/silica under mechanochemical conditions to afford products 159a in good to excellent yields (81-94 %). Then, toward series b, same starting materials were combined but under mechanochemical and solventless conditions to synthesize products 159b in moderate to excellent yields (35-99 %). Both series were obtained in a good diastereomeric ratio (up to 2:1). Aromatic and aliphatic aldehydes with releasing and withdrawing groups were used. Also, it is noteworthy the role of silica as a dehumidifier and partial-acidic agent at the grinding time.
Ismaili et al. reported the synthesis of many new heterocyclic compounds via Biginelli reaction which were evaluated toward three different biological targets [37]. Thus, the key substrates were the alkynyl-aldehydes 163, which were synthesized previously through two routes: a) Williamson reaction between 4-hydroxybenzaldehyde (160) and propargyl bromide (161), and b) 4-hydroxybenzaldehyde (160) and alcohol 162 via a Mitsunobu reaction (Scheme 25). On the other hand, ureas 167 were prepared from benzylpiperidines 164 and benzoyl isocyanate or benzoylthioisocyante (165). Thus, aldehydes 163, ureas 167 and ethyl acetoacetate (168) furnished the compounds 169 in moderate yields (10-39 %) making use of sodium bisulfate as catalyst. These kinds of compounds have potential activity as Ca2+ channel blockers, cholinesterase inhibitors, and activators of nuclear erythroid 2-related factor 2.
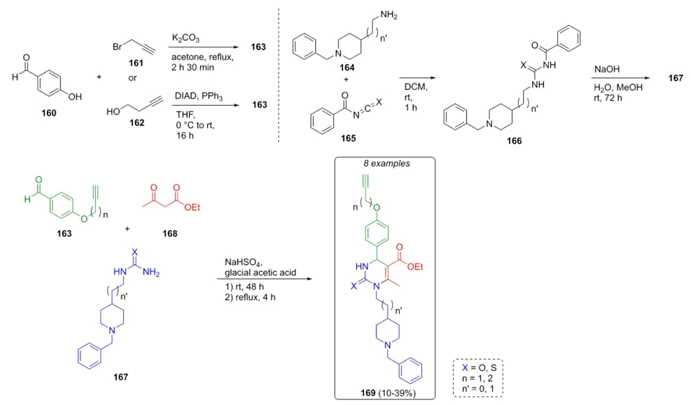
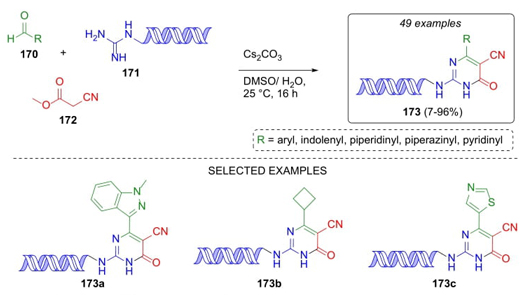
Qi, Peng et al. developed a innovative strategy in which DNA-derivatives were obtained [38]. After optimization of reaction conditions, aromatic and heterocyclic aldehydes 170, DNA-conjugated guanidine 171, and methyl cyanoacetate (172) were sequentially combined using cesium carbonate as the base to achieve DNA-dihydropyrimidinones 173 in low to good yields (7-96 %) (Scheme 26). Thus, isocytosine scaffolds were developed taking advantage of DNA-conjugated guanidine fragment. A variety of electron-donating and releasing groups were tolerated in such reactions even 5-membered heteroaromatic compounds participated therein. It is important to highlight that this is the first report in which a DNA-amine fragment was used in an organic reaction.
Reissert-type 3CRs
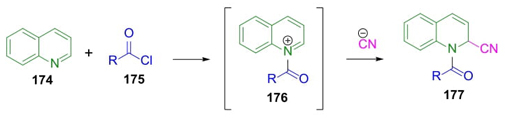
Cooney’s manuscript from 1982 described that Arnold Reissert reported in 1905 a multicomponent reaction involving combinations of quinoline with acyl chlorides, and potassium cyanide in aqueous media (Scheme 27) [39]. Thus, the formation of N-acyl quinoliniums 176 by the reaction of quinoline (174) and acyl chlorides 175 is followed by cyanide anion addition to promote the formation of the Reissert products 177. The preparation of such compounds in aqueous media has some common complications. For instance, the low solubility of the reagents because of their different polarity that may be solved by using mixtures of solvents. Reissert reaction works well on a wide range of pyridine-containing reagents, acyl chloride-derivatives, and different kind of nucleophiles [40].
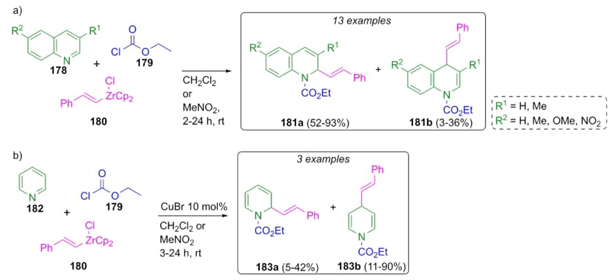
Reissert-type reactions have been developed using a variety of nucleophiles instead of cyanide anions. Thus, Saito, Hanzawa et al. reported a methodology where alkenylzirconocene chloride was used as a nucleophile in a Reissert-type 3CR (Scheme 28(a)) [41]. This synthetic methodology consisted of a sequential combination of quinoline derivatives 178 or pyridine (182), styrylzirconocene 180 and ethyl chloroformate (179) in dichloromethane or nitromethane to give quinoline- or pyridine-containing products 181 or 183, respectively, in moderate to good yields (up to 93 %). The use of nitromethane instead of dichloromethane as the solvent improved the 1,2-additions on the quinoline derivatives. However, the use of nitromethane in the reaction of pyridine afforded a mixture of regioisomers with a poor 1,2-addition selectivity (Scheme 28(b)). On the contrary, the 1,4-addition was improved by employing a catalytic amount of CuBr (10 mol%) at -78 °C to the pyridine, whilst using quinoline derivatives irrelevant selectivity was found (Scheme 28).
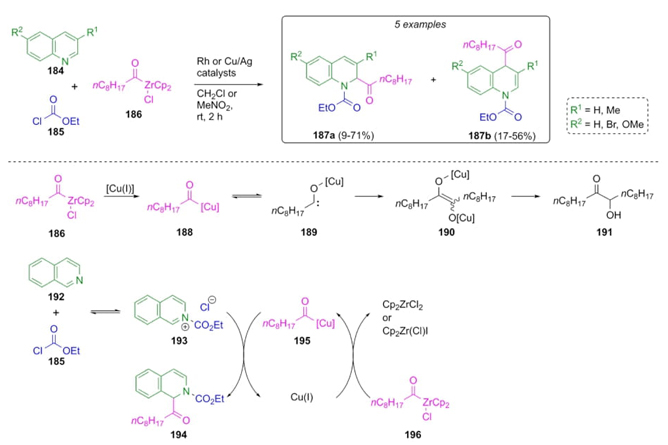
Later in 2013, Saito et al. continued developing a similar strategy, finding that the orientation of the reaction can be controlled by the addition of copper or rhodium catalysts (Scheme 29) [42]. Thus, a sequential combination of polysubstituted quinolines 184, acylzirconocene chloride (186), and ethyl chloroformate (185) under copper iodide (I) or rhodium/silver catalysts afforded a mixture of regioisomeric products 187 in moderate to good yields (up to 71 %). A mechanism pathway was proposed where chloride 186 was coordinated with Cu(I) to give the complex 188 which performed compound 191 after the formation of carbene 189 and its precursor 190. Thus, there is an equilibrium between the addition of isoquinoline (192) and chloride 185 with N-acylisoquinolinium 193 and acylated product 194. Acyl cooper product 195 was carried out through acyl transfers from the chloride 196 to Cu(I). In the case of the combination of rhodium (2.5 mol%) and silver (5 mol %) catalysts, it was promoted 1,2-addition in nitromethane as solvent. Instead that, under copper catalyst (10 mol %), 1,4-addition is promoted in dichloromethane. Then, N-acylation of isoquinoline occurred at the 2-position through an addition of the corresponding nucleophile.
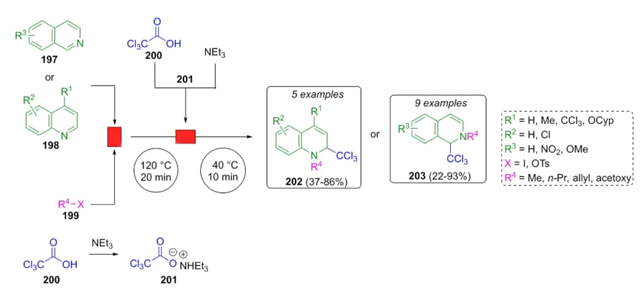
Lindhardt et al. developed a methodology to obtain isoquinolines and quinolines via a decarboxylative Reissert-type reaction through a continuous flow [43]. Isoquinolines 197 or quinolines 198, triethylammonium trichloroacetate (201), and electrophilic iodides or tosylates 199 were combined in neat DMF or a mixture of DMF/ MeCN under continuous flow afforded dihydroquinolines 202 or dihydroisoquinolines 203 in moderate to good yields (up to 93 %) (Scheme 30). N-alkylations were carried out with different types of alkylic chains. 4-Addition was promoted by trichloromethyl anion to afford the products 202. On the other hand, alkyl iodides or allyl tosylates promote the corresponding alkylations to give the products 203.
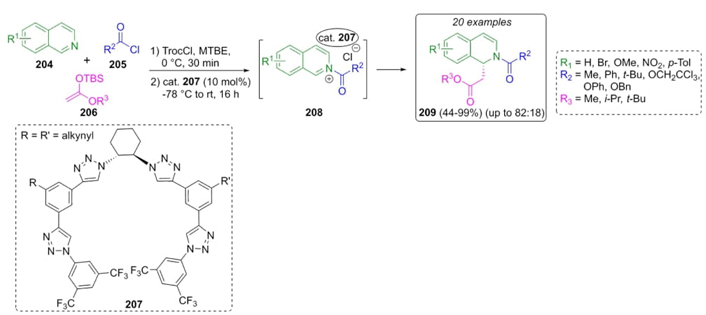
García-Mancheño et al. used a chiral triazole-based organocatalyst to synthesize some new Reissert-type dihydroisoquinolines (Scheme 31) [44]. After optimizing reaction parameters, a sequential combination of isoquinolines204, acid chlorides 205, and silyl ketene acetals 206 in methyl tert-butyl ether (MTBE), catalyzed by the triazole-containing organocatalysts 207 resulted in the synthesis of isoquinolines 209 in moderate to good yields (up to 99 %) and good enantioselectivity (up to 82:12 (R/S). The use of chiral-triazole through anion-binding activation and MTBE as solvent had a strong influence on the enantioselectivity. 1H-1,2,3-triazoles are excellent H-bond donors due to the high polarizability of their C-H bond and the acidity at the C-5 position. In addition, triazole catalysts exhibit chloride counterion binding, which results in chirality transfer. This type of catalysis is most efficient in the case of counterion catalysts with halogen-halogen interactions [45].
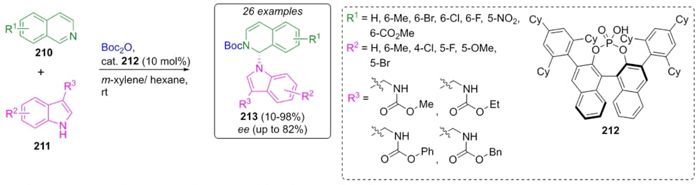
You et al. developed an asymmetric N-alkylation of indoles via a Reissert reaction (Scheme 32) [46]. Thus, once the optimal reaction conditions were identified, a sequential combination of isoquinolines 210, indoles 211, and protective group reagent Boc2O catalyzed by the chiral phosphonic acid 212 in the solvent mixture (m-xylene/hexane) at room temperature allowed the synthesis of compounds 213 in good to excellent yields (up to 98 %) and with moderate to good enantioselectivities (up to 82 %) as (R) absolute configuration. Indole alkylation occurred in the N-1 position assisted by the catalyst. The best yields and enantioselectivities were obtained when the reaction occurred at position 2 of the isoquinoline ring, which is due to steric hindrance at the other reactive positions.
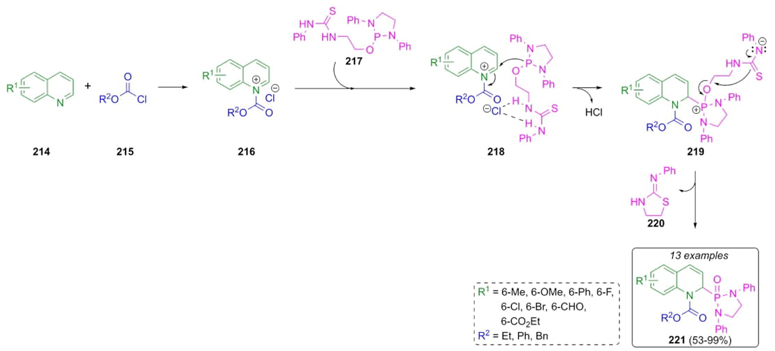
Kang et al. reported the synthesis of a series of new quinolinyl phosphonamides via a Reissert-type reaction (Scheme 33) [47]. A plausible mechanism involves the formation of quinolinium salts 216 through a mixture of quinolines 214 and chloroformates 215. Thus, thiourea 217 reacts with quinolinium salts 216 to give the salt-complexes intermediates 218. Finally, an intramolecular nucleophilic attack on intermediates 219 occurs to release cyclic compound 220, achieving the products 221 in moderate to good yields (53-99 %). The salt-complexed thioureas 218 as Brønsted acids promote 1,2-regioselective additions of the phosphine moiety through a ion paring process between chloride and thiourea fragments.
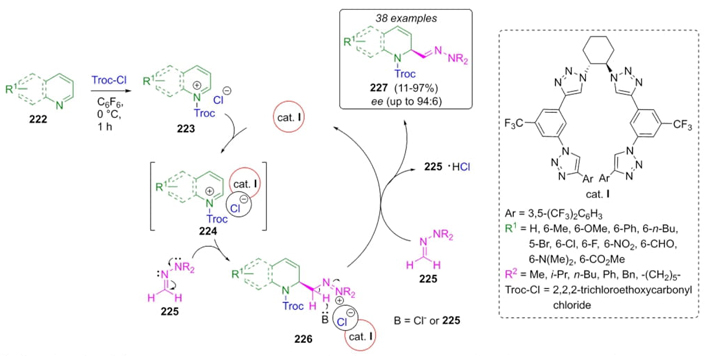
In 2021, García-Mancheño et al. developed a robust strategy to synthesize 2-hydrazone-1,2-dihydroquinolines 227 via a Reissert-type reaction using a triazole-containing organocatalyst and dimethylhydrazone as the nucleophile with reversed-polarity [48]. The best reaction conditions were employing CF3-tetrakistriazole as the catalyst and hexafluorobenzene as the solvent at low temperatures. Thus, quinolines 222, trichloroethoxycarbonyl chloride (Troc-Cl), and formaldehyde dimethylhydrazones 225 were reacted together to give the products 227 in moderate to good yields (11-97 %) and enantiomeric ratio (er) up to 94:6 (Scheme 34). It was proposed a plausible mechanism in which quinolinium salts 224 and 226 were formed after a chloride activation with Troc-Cl to give intermediates 223. Then, the later were activated through an anion-binding catalysis. Finally, a nucleophilic attack of the hydrazones followed by deprotonation afforded products 227. It is to highlight that the use of hexafluorobenzene as the solvent and tetrakestriazole as the organocatalyst promoted a high regio- and chemoselectivity through anion-binding strategy; which is probably due to the presence of triazoles-CF3 moieties as a key fragment in the organocatalyst, as discussed above.
Mannich-type 3CRs
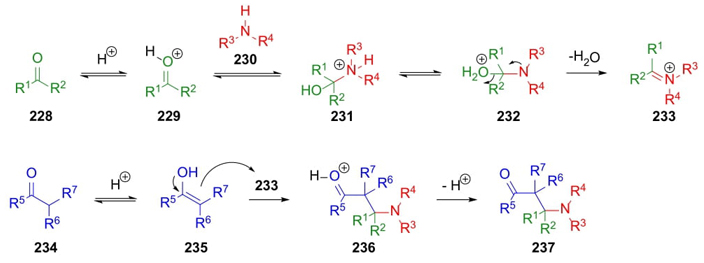
Around 1917, Carl Mannich reported a 3CR that consists of the combination of a non-enolizable carbonyl compound (usually aldehyde or ketone), a primary or secondary amine (even ammonia), and an enolizable aldehyde or ketone to access β-amino carbonyl compounds. A mechanism accepted begins with the formation of oxoniums 229 through protonation of carbonyl compounds 228. Then, addition of amines 230 to give species 231 which after prototropic exchange achieve intermediates 232, and the formation of iminium ions 233 through water release. Besides, carbonyl compounds 234 perform corresponding enols 235. Then, a nucleophilic attack from enols 235 to iminium ions 233 achieved oxonium salts 236. Finally, a proton releases complete formation of alkaloid-type compounds 237(Scheme 35) [49]. This procedure can also be carried out as intramolecular manner [50]. The asymmetric version of this process could be catalyzed by organic and inorganic complexes. This reaction has been applied successfully for the synthesis of several natural products, even some alkaloids [51].
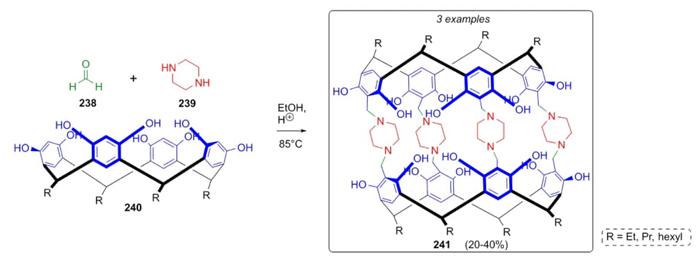
Rissanen et al. developed a one-pot synthetic approach based on the Mannich reaction to synthesize three complex macrocycles 241 [52]. Formaldehyde (238) in solution of ethanol was combined with piperazine (239) and cyclic tetrabenzoxazines 240 to assemble the macrocyclic dimers 241 in modest yields (20-40 %) (Scheme 36). Piperazine links both moieties through covalent bonds giving rigidity to these supramolecules. Despite the size of the compounds, the yields were moderate highlighting that the one-pot process was favored by the solvent being encapsulated on cage structures.
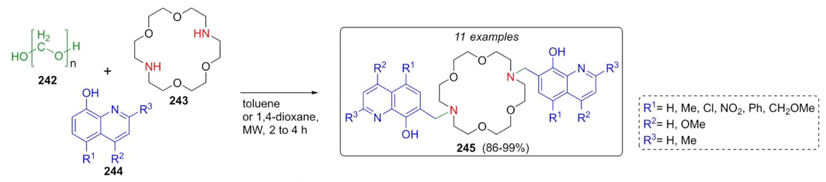
Aza crown-ether-derivatives have been utilized as fluorescent sensors in different biological targets. Lombardo, Trombini et al. synthesized eleven new diaza crown-ethers via a Mannich one-pot process using microwave radiation as a heat source [53]. Paraformaldehyde 242, and the crown-ether 243 were combined sequentially with 8-hydroxyquinolines 244 reaching ligands 245 in good yields (86-99%) via a microwave-assisted Mannich-type 3CR (Scheme 37). It is important to highlight the high atom economy of this innovative experimental protocol. Also, microwave-assisted catalyst-free conditions decreases the formation of by-products. Strategic substituents at positions 2, 4 and 5 in isoquinoline moiety contributed to higher yields due to a high activation on such ring.
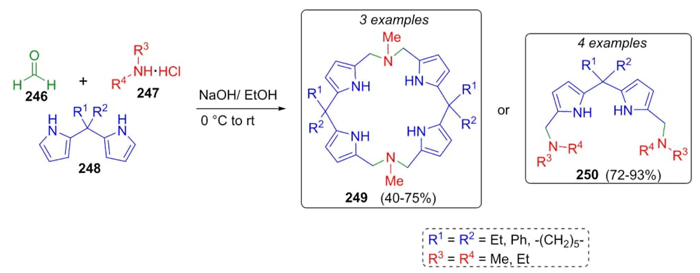
Mani et al. reported the synthesis of a series of diazacalix[2]dipyrrolylmethanes (macrocyclic 249 and acyclic 250) via a Mannich 3CR from formaldehyde (246), amine hydrochlorides 247, in the presence of dipyrrolylmethane-derivatives 248 in moderate to good yields (40-93%) (Scheme 38) [54]. It is exemplified by another three-component approach where flexible anion-receptor supramolecules were assembled. Also, these reactions were carried out under catalyst-free conditions. These kinds of compounds by containing both hydrogen donors and acceptors can act as ion-pair receptors or anion receptors, which is important mainly in medicinal chemistry.
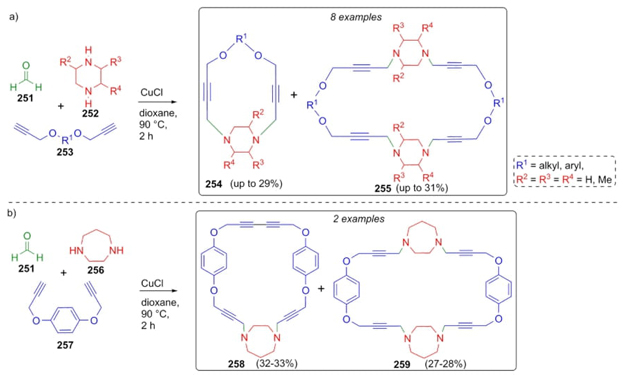
Coined for the first time by C.-J. Li, the A3 coupling is a 3CR of amines, aldehydes, and alkynes to synthesize propargyl amines. [55]. This coupling, which is characterized by its high atom economy and ecofriendly conditions, is commonly applied for the formation of several kind of N, O, and S-heterocycles [56]. In 2011, Wu et al. reported an interesting methodology to generate macrocycles via a Mannich reaction based on an early A3 coupling [57]. Thus, a copper(I)-catalyzed sequential combination of formaldehyde (251), polysubstituted piperazines 252, and dialkynes 253 gave the macrocycles 254 and 255 in moderate yields (29-31 %) (Scheme 39(a)). In addition, another Mannich-type 3CR coupled to an Eglinton-Glaser dimerization was performed (Scheme 39(b)) by using formaldehyde (251), 1,4-diazepane (256), and aryl-dialkynes 257 to give the macrocycles 258 and 259. Both terminal dialkynes 253 or 257 fulfilled the double function in the Mannich reaction and dimerization process. Also, these kind of reactions proceed correctly using secondary amines, which is not common.
In 2015, Singh et al. reported an interesting procedure to synthesize a new series of isoindolinones and tetrahydroisoquinolines via a domino Mukaiyama−Mannich lactamization/alkylation process [58]. Thus, to synthesize the isoindolinones 264, the methodology was optimized using metal triflates as catalysts and electron-donor-acceptor substituted benzaldehydes 260 and amine 261, finding that electron-withdrawing amines do not promote such reaction. In the same way, o-anilines do not allow the corresponding isoquinoline formation due to steric hindrance (Scheme 40(a)). Thus, the same strategy was improved to assemble the corresponding tetrahydroisoquinolines 269. Thus, the combination of aldehydes 265, amines 266, and silyl enol ether- derivatives 267 catalyzed by Lewis acids achieved the products 269 in moderate to good yields (65-88 %) (Scheme 40(b)). To note, a decrease in reaction time was found for the synthesis of tetrahydroisoquinolines 269 probably due to a higher reactivity of intermediates 268 in comparison to intermediates 263.
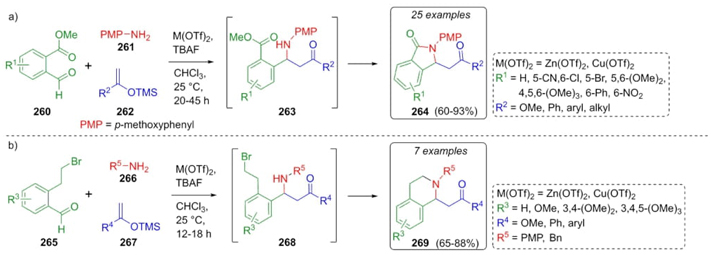
Scheme 40 Synthesis of isoindolinones and tetrahydroisoquinolines via metal-catalyzed Mannich-type 3CR.
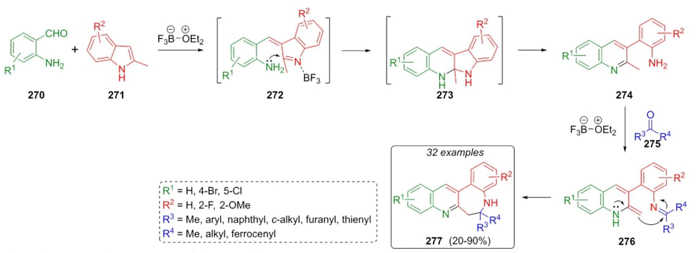
Gu et al. reported a procedure to synthesize the quinoline-fused 1‑benzazepines 277 via an intramolecular Mannich 3CR [59]. Thus, o-aminobenzaldehydes 270, indoles 271 as the nucleophile/amine-containing components, and ketones 275 were sequentially combined using a Lewis acid catalyst to give the benzo-annulated species 277 in moderate to good yields (20-90 %) (Scheme 41). The most plausible reaction mechanism involves the formation of intermediates 272 followed by nucleophilic attack at C2 position on the indole ring to give cyclized intermediates 273. Then, a ring-opening achieved compounds 274. Finally, compounds 277 were achieved through intramolecular cyclizations of activated enamines 276. A wide range of electron-donating and electron-deficient groups was included in the substrate scope. Even bulky groups such as ferrocene and cyclooctanone gave moderate yields. Formation of C, N-bis nucleophile intermediates 274 through a combination of aldehydes 270 and indoles 271 was the key step in this strategy.
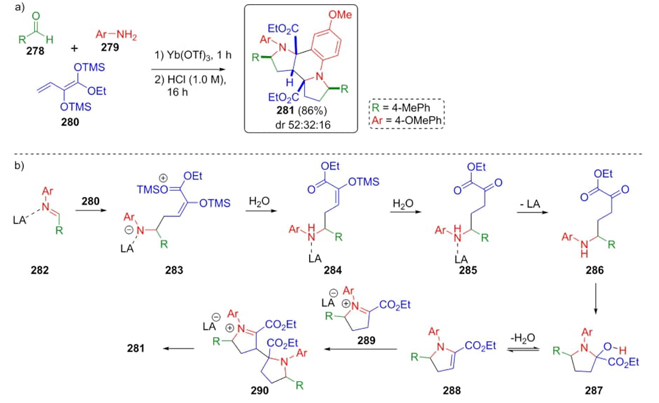
In 2018, Schneider et al. developed a synthetic procedure to obtain dipyrroloquinolines via a Mukaiyama−Mannich reaction [60]. The strategy involved a key mixture between the Lewis-acid activated imines 282 and a bis(silyl) dienediolate 280, which after a few steps gave the products 281 (Scheme 42). To carry out this synthesis, it was achieved a three-component reaction through sequential combination of aldehydes 278, amines 279, and TMS-dienediolate 280 under catalysis with Ytterbium (III) in acid medium to give a diastereomeric mixture (52:32:16) of products 281 in good chemical yield (86 %). The Lewis-acid catalyzed imines 282 and dienediolate 280 furnished desilylated enol ethers 283which after the addition of water performed compounds 284 and 285 and release of Lewis acidafforded the keto esters 286. Then, it was promoted an intramolecular cyclization to furnish compounds 287 in equilibria with compounds 288. Finally, it was carried out the Mannich reaction followed by Pictet-Spengler cyclization. In this strategy, several C-C and C-N bonds were formed with excellent stereoselectivity using both Brønsted and Lewis acids, where Brønsted acids enhanced better the stereoselectivity. DFT-based calculations by using the functional M06-D3 at LACVP**/PBF level of theory support the proposed mechanism.
Abe, Yamada et al. developed a robust methodology to synthetize polyheterocycles via a ring-opening/vinyl closure cascade strategy [61], involving a key vinylogous Mannich/retro Mannich process (Scheme 43). Thus, indoline-salts 291, carbolines 293, and triethylamine reacted together in a one-pot fashion to assemble the polyheterocycles 298 in moderate to excellent yields (32-95 %). A plausible mechanism was proposed where epoxides 292 were achieved from salts 291 and triethylamine, followed by the formation of ammonium salts 294 through addition of indols 293 to epoxides 292. Then, an ammonium exchange promotes a Hoffman elimination to give intermediates 295, followed by a vinylogous Mannich reaction between carbolines 293 and intermediates 295 to afford alkylated intermediates 296. A retro-Mannich process achieved intermediates 297. Finally, aintramolecular cyclization on the intermediates 297 complete the synthesis of compounds 298. It is worth mentioning the absence of catalysts in all cascade processes, and the high atom economy obtained. Electron-donating and electron-deficient groups had a strong influence on the yields, especially substituents at the C-5 position of the indole and piperidine rings. On the other way, electron-withdrawing groups placed at the C-6 on the carbolines caused the lowest yields.
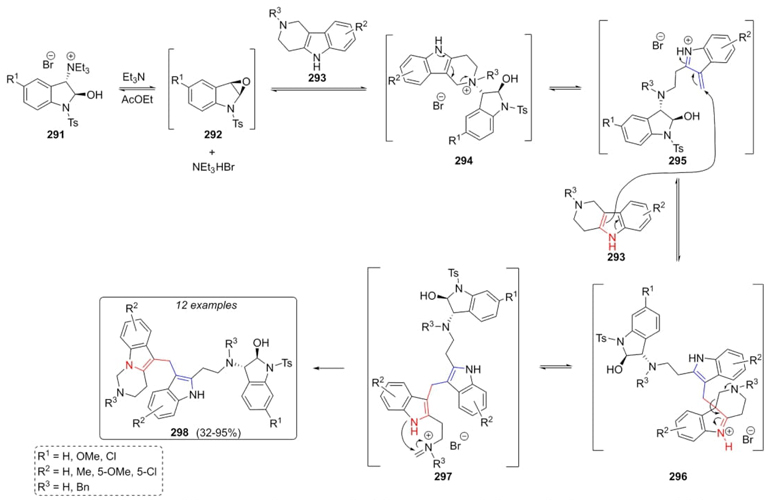
Scheme 43 Synthesis of polyheterocycles via a double ‘open and shut’ vinylogous Mannich/retro Mannich process.
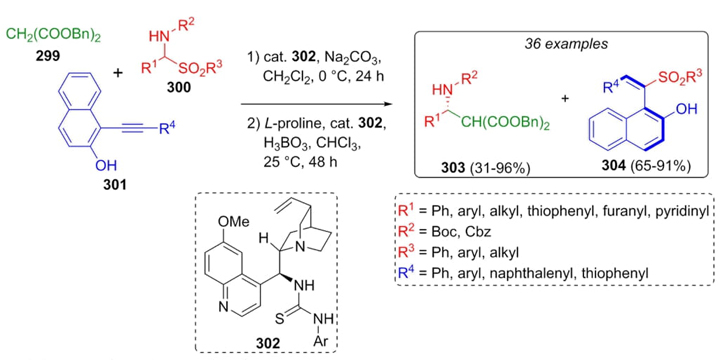
Yan et al. reported an interesting method involving a waste and reuse-based tandem Mannich reaction (Scheme 44) [62]. Amido sulfones 300, activated-methylene compound 299, and alkynes 301 reacted together under organocatalytic and mild basic conditions affording both chiral products, the b-amino acid-derivatives 303 (asymmetric compounds) and naphthyl-styrenes 304 (rotation-restricted naphthalene-type enantiomers) in moderate to excellent yields (31-96 % and 65-91 %, respectively). Thus, good enantioselectivity for each asymmetric product was found. Besides, a wide variety of electron-withdrawing and electron-releasing groups were efficiently employed in Mannich 3CR through a waste-reuse process to afford at the same time sulfone-containing naphthyl-styrenes and amino acid derivatives.
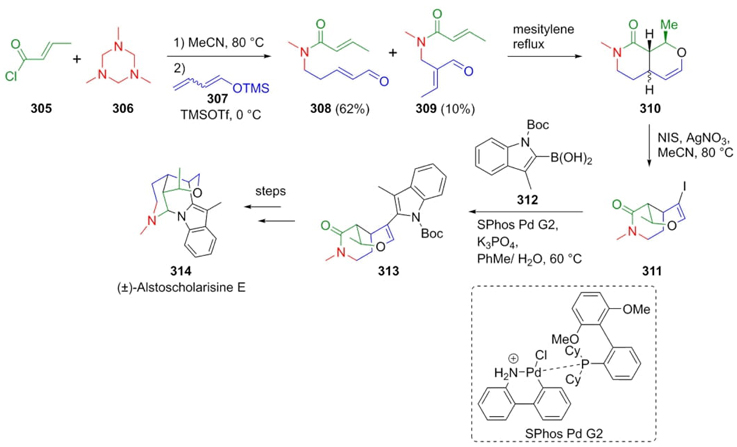
Martin et al. achieved the total synthesis of (±)-Alstoscholarisine-E (314) in seven steps with high stereoselectivity employing a vinylogous Mannich-type reaction as the initial key process [63]. Thus, acid chloride 305 and triazine 306 were combined in a solution of acetonitrile at 80 °C affording the in situ formed N-acyliminium ion, to which protected diene 307 was added to provide regioisomeric amides 308 and 309 in 62 % and 10 % yield, respectively (Scheme 45). Subsequent transformation of such compounds included a hetero-Diels-Alder cycloaddition, stereoselective enol ether reduction, a Suzuki-coupling, and a Lewis acid-catalyzed reduction. The three-component vinylogous Mannich reaction had a key role in this creative and short total synthesis of the natural product 314.
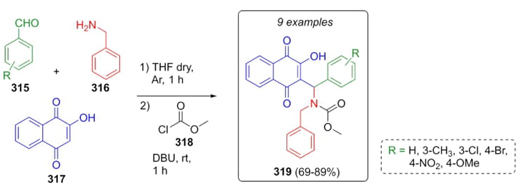
Forezi, Robbs et al. reported the synthesis of nine new naphthoquinones via a Mannich one-pot reaction, and their evaluation against oral squamous cell carcinoma was conducted [64]. Aromatic aldehydes 315, benzylamine (316), and hennotannic acid (317) also known as lawsone were combined sequentially at room temperature (Scheme 46). Then, DBU and methyl chloroformate (318) were added to obtain the naphthoquinone derivatives 319 in moderate to good yields (69-89 %). Also, dialkylated products were also obtained but changing the base by K2CO3. Naphthoquinones reported exhibited potential activity for preclinical trials against oral squamous cell carcinoma.
Willgerodt-Kindler-type 3CRs
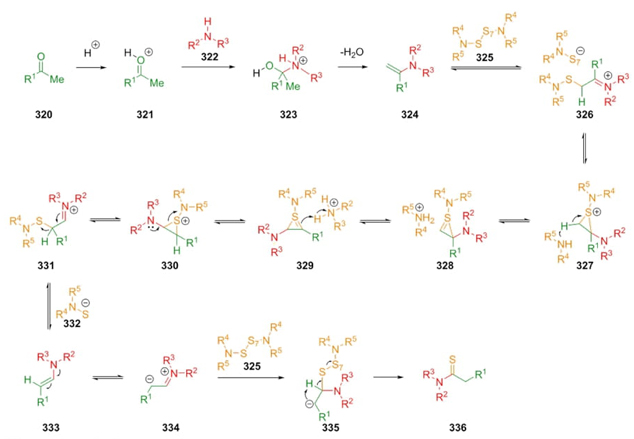
In 1887 (C. Willgerodt) and in 1923 (K. Kindler) the reaction between carbonylic compounds, disulfide compounds (or elemental sulfur), and secondary amines was developed, and later named asWillgerodt-Kindler reaction. In its classical version, this kind of one-pot process involves combinations of a carbonylic compound (aldehydes or ketones), elemental sulfur, and amines. Other non-carbonylic (or unsaturated) reactants such as imines, amines, alkenes, alkynes, or acetals may be used in these kinds of reactions [65]. In 1989, M. Carmack reported the mechanism in which the main steps consisted in the formation of enamines 324 between ketones 320 and amines 322 in acid media. Then formation of iminium ions 326 through reaction of enamines 324 and amino-sulfur compounds 325. Thus, it were performed annulated intermediates through a set of thiirene-type rearrangements 327 to 330 to achieve enamines 331.Finally, an oxidation on compound 333 in equilibria with 334 promoted by sulfur-containing reagents 325 afforded the products 336 (Scheme 47) [66].
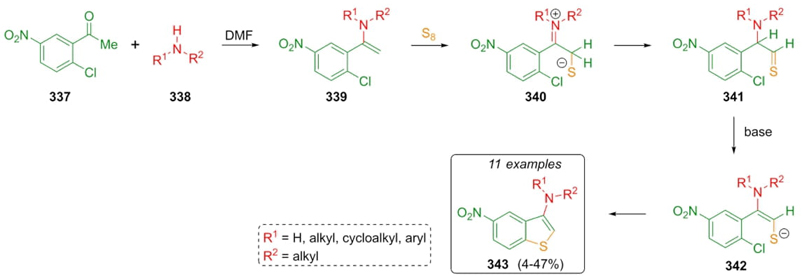
Androsov et al. reported a strategy based on the Willgerodt-Kindler reaction to synthesize 3-amino benzo[b]thiophenes. Thus, acetophenone 337, amines 338, and elemental sulfur were mixed obtaining the products 343 in modest to good yields (4-47 %) [67]. A plausible mechanism of reaction was proposed (Scheme 48). Enamines 339 were formed by condensation of ketone 337 and amines 338, and the sulfur-was added to give the intermediates 341. Finally, the ring closure step took place by a nucleophilic substitution on intermediates 342 assisted by the nitro group, which activates its para-position substituted by the chlorine atom. Yields may be affected by the steric hindrance of the secondary bulky amines and low temperatures.
In 2013, Eftekhari-Sis, Büyükgüngör et al. reported the first Willgerodt-Kindler reaction using arylglyoxals as carbonylic reagents. Thus, phenylglyoxals 344, amines 345, and elemental sulfur were combined to achieve products 350 in good to excellent yields (70-90 %). It was proposed a plausible reaction mechanism where a pathway involving an iminium ion was followed instead of one involving formation of enamines. Thus, it was proposed the classical mechanism. Iminium ions 346 were formed from glyoxals 344 and amines 345. Zwitterions 348 were performed through intermediates 347. Then, the addition to iminium ions 346 to iminium ions 348 furnished the intermediates 349, which after a release of protonated amine performed the products 350 (Scheme 49) [68]. It is important to underline the high efficiency obtained under solventless conditions. Also, the reaction worked well using aryl-glyoxals with electron donator and deficient groups, as well as using secondary cyclic amines. On the other hand, the reaction did not proceed with primary amines or nitro-containing glyoxals.
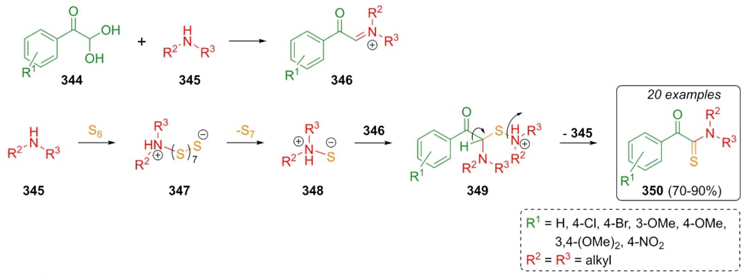
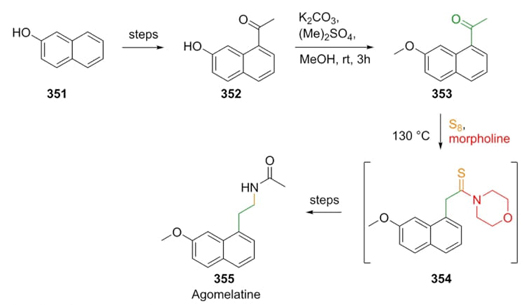
Cherukupally et al. furnished the total synthesis in eight steps of the antidepressant drug agomelatine (355) by a synthetic strategy in which the key step was a Willgerodt-Kindler reaction [69]. The procedure began with 2-naphthol 351, which was transformed into ketone 352. The latter was methylated with dimethyl sulfate in mild basic conditions to give ketone 353. Then, ketone 353, morpholine, and elementary sulfur were converted into thiomorpholide-type intermediate 354 (Scheme 50). Further transformations such as esterification, reduction to alcohol, and hydrogenation completed the synthetic route in 27 % overall yield. The use of a Willgerodt-Kindler reaction in a total synthesis, since this process regularly provides moderate to good yields.
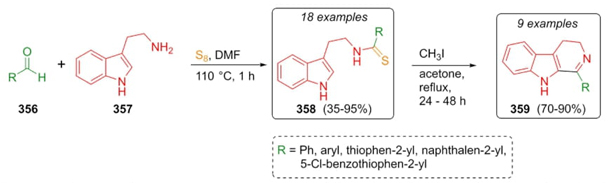
Volk et al. performed the first Willgerodt-Kindler 3CR-based synthetic strategy towards N-thioacyltryptamines 358, and their further transformations into the corresponding β-carbolines 359 (Scheme 51) [70]. Thus, aldehydes 356, tryptamine (357), and sulfur were combined to afford the N-thioacyltryptamines 358 in moderate to good yields (35-95 %). At the same time, a portion of these products were carried to subsequent ring-closure transformations promoted by iodomethane to give β-carbolines 359. In addition, this procedure was furnished using a broad range of electron-releasing and electron-withdrawing groups placed in the aldehydes 356.
In 2017, Darehkordi et al. performed a rapid route to synthesize novel heterocycles incorporating fragments of antibiotic fluoroquinoline-derivatives such as enoxacine, norxacine, and ciprofloxacine via a Willgerodt-Kindler reaction [71]. Thus, arylglyoxals 365, fluoroquinolones 363, and elemental sulfur were combined sequentially using sodium sulfide in catalytic amounts to reach products 369 in good to excellent yields (83-95 %). The mechanism proposed suggests that Na2S (360) and sulfur (361) allowed the formation of ions 362 that combined with amines 363 gave the anions 364, which perform a nucleophilic attack on iminium ions 367 to achieve the products 369 through a release of amines 363 from the intermediates 368 (Scheme 52). It is worth emphasizing the key role of Na2S (360) in the addition to uncommon amine source and the role of sulfur as an oxidant. Also, this one-pot reaction allowed easily and efficiently incorporation of fluorodihydroquinoline as an antibiotic pharmacophore fragment.
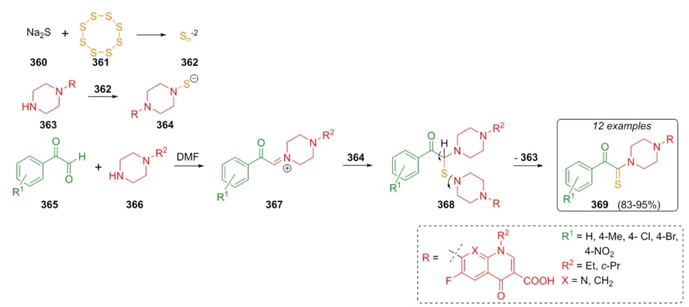
Scheme 52 Synthesis of piperazine-linked fluorodihydroquinoline-3-carboxylic acids with potential antibiotic activity.
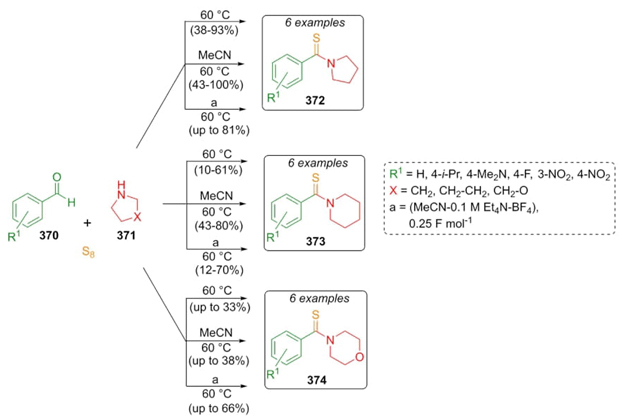
Feroci et al. reported a sustainable method to synthesize thiobenzamides via a Willgerodt-Kindler reaction [72]. Benzaldehydes 370, cyclic amines 371, and elemental sulfur were sequentially combined in three different reaction pathways: under solventless conditions, in solution, and by electrochemical conditions at the same temperature (60 °C) for 3 h to achieve products 372, 373, and 374 (Scheme 53). The yields were varied (up to qualitative yields) due to the variety of substituents in the aromatic aldehydes. Regarding the electrochemical reactions, it is not easy to carry out this type of reactions with electro-attracting groups, but in the case of this methodology, it worked satisfactorily. Also, in electrochemical conditions, formation of tetraethylammonium cations was observed. Low yields were obtained for morpholine-containing analogues probably due to the interactions between oxygen from the morpholine moiety and the sulfur source even though stoichiometric amounts of sulfur were used at low temperatures.
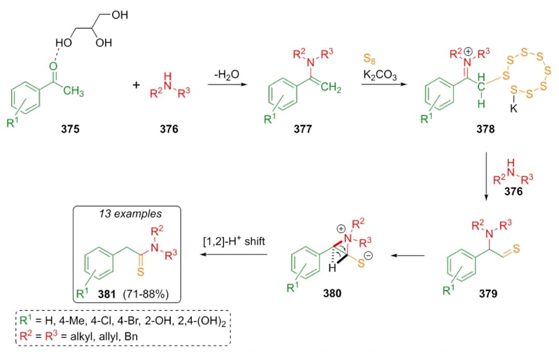
In 2017, Heydari et al. developed a green approach to synthesize ethanethioamides under a basic deep eutectic solvent via the Willgerodt-Kindler reaction [73]. After optimizing solvent conditions, thioamides 381 were obtained in good yields (71-88 %) by sequential combination of acetophenones/glycerol 375, amines 376, and sulfur under glycerol/carbonate-assisted solvent. A plausible mechanism was described in Scheme 54, where glycerol plays an important role in capturing water producing the corresponding enamines 377. Then, the base promoted the formation of sulfur anion and deprotonation of iminium ions 378, which after the addition of amines 376 achieved compounds 379 that through a rearrangement afforded intermediates 380, and a 1,2-proton shift performed the products 381. Moreover, the deep eutectic solvent, glycerol-K2CO3, increases collisions in the reaction medium promoting a better chemical environment.
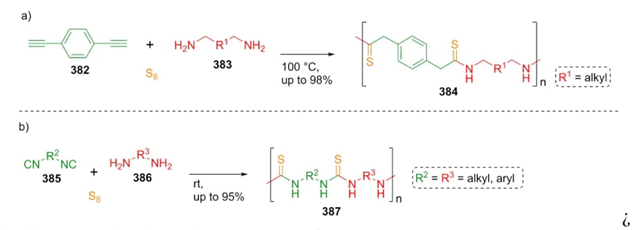
In 2018, Theato et al. published a seminal manuscript in which two types of multicomponent polymerizations via a Willgerodt-Kindler reaction are described [74]. Reaction a) combining dialkynes 382 with aliphatic diamines 383 , and reaction b) using diisonitriles 385 and aliphatic diamines 386. Both reactions were carried out under typical Willgerodt-Kindler conditions and without catalysts, yielding up to 98 % and 95 % for both kind of polymeric products 384 and 387 respectively (Scheme 55).
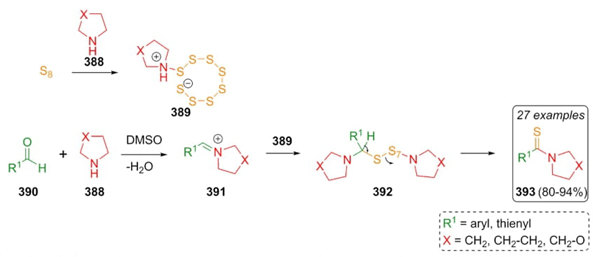
In 2019, Dalal et al. developed a methodology to synthesize thioamides via a Willgerodt-Kindler reaction [75]. Thioamides 393 were assembled by combining aromatic aldehydes 390, cyclic amines 388, and a sulfur source under catalyst-free conditions. The products were prepared in good to excellent yields (80-94 %). Electron-withdrawing and electron-donating groups were included in the substrate scope. A plausible reaction mechanism starting from zwitterionic compounds 389, which were prepared by reacting sulfur and amines 388. Then, zwitterions 389 performed a nucleophilic attack on cyclic iminium ions 391 to give intermediates 392 (Scheme 56). Finally after releasing of sulfur, the products 393 were achieved. The best yields were directed by three important factors: a) nucleophilicity of cyclic secondary amines; b) DMSO as an environmentally friendly solvent; and c) low amounts of sulfur used.
In 2019, Huang, Ji et al. reported an interesting 3CR approach which includes a cascade process under transition metal-free conditions, and employing the Willgerodt-Kindler-type reaction [76].Polyheterocyclic thiazoles 404 were synthesized up to 82 % yield through sequential combinations of ketoximes 396, isothiocyanates 403, and a sulfur source promoted by two bases. A plausible mechanistic pathway suggests formation of a base-promoted sulfur radicals 394 s, which are inserted into ketoximes 396 followed by a [1,3]-H shift in the intermediates 397 to give radicals 398. Then, it was promoted a radical cyclization on compounds 398 to achieve intermediates 399, which after a single electron transfer (SET) performed heterocycles 400, and finally, a Willgerodt-Kindler-type reaction between thiols 402 and isothiocyanates 403 (Scheme 57). Notability, sulfur-assisted cascade process formed trisulfur radical anion 394, which activated compound 396. The base-containing acetates promoted the formation of subproducts. Ketoxime acetates proceed accurately when they are substituted in meta instead of ortho position. On the other hand, other reagents were explored instead of isocyanates such as urea, thiourea, DMF, etc. but those attempts did not proceed.
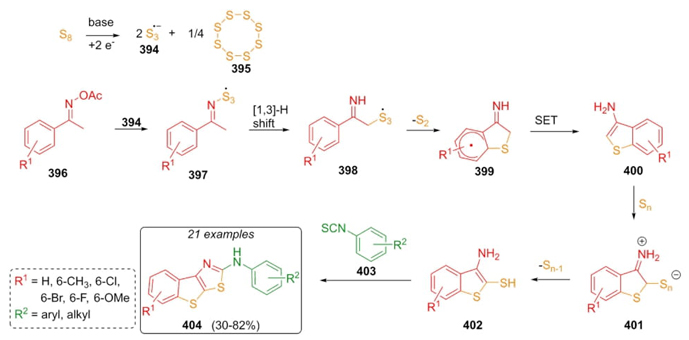
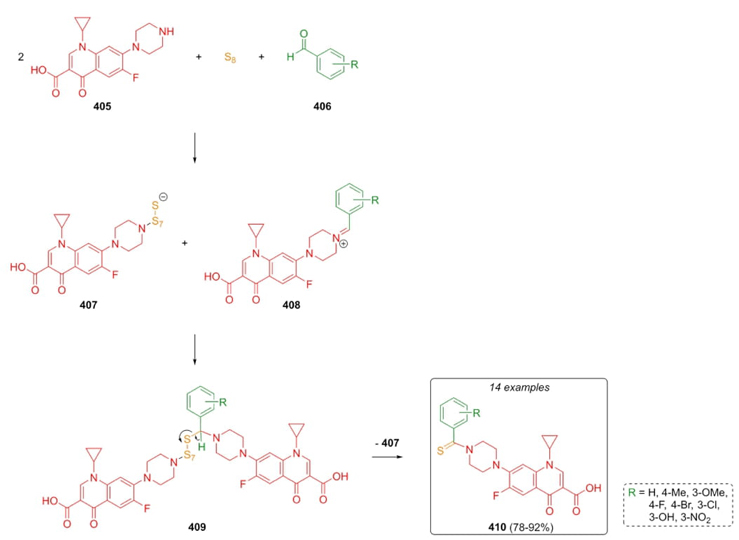
In 2022, Iraji, Mahdavi et al. developed a synthetic procedure to achieve ciprofloxacin derivatives through a Willgerodt-Kindler-3CR, incorporating a pharmacophoric fragment behind the search for potential antibacterial activity [77]. The combination of aromatic aldehydes 406, a couple of equivalents of ciprofloxacin (405), and sulfur allowed the synthesis of ciprofloxacin derivatives 410 in moderate to good yields (78-92 %). A plausible mechanism was proposed (Scheme 58). Condensation of aldehyde and amine-fragment from ciprofloxacin performed iminium ions 408. Besides, nucleophilic attack from sulfur to ciprofloxacin (405) gave the polysulfide anion 407. Finally, a reaction between iminium ions 408 and anions 407 after an oxidation process, ciprofloxacin derivatives 410 were obtained via elimination of ciprofloxacin anion 407 from the precursors 409. It is worth noting that a modest to good antibacterial activity was exhibited by compounds 410 with the ciprofloxacin pharmacophoric fragment included.
Bücherer-Bergs-type 3CRs
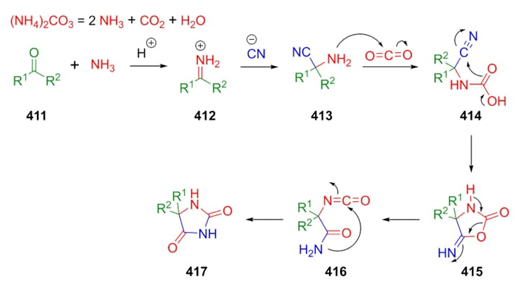
In 1934, H. T. Bucherer reported a mechanism for hydantoin formation which was previously patented by H. Bergs in 1929 [78]. Thus, Bucherer-Bergs reaction is a multicomponent reaction between carbonylic compounds (aldehydes or ketones), a cyanide source (KCN), and ammonium carbonate. The general reaction pathway involves the formation of imines 412, which are attacked by cyanide anions to afford the corresponding aminonitriles 413 followed by a nucleophilic addition to CO2 to furnish intermediates 414. A ring-closing process occurs to give the cyclic compounds 415, which undergo a rearrangement toward formation of isocyanate-type intermediates 416 to finally furnish the hydantoin-based compounds 417 (Scheme 59) [79].
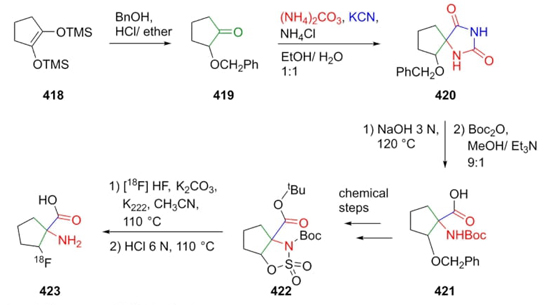
Goodman et al. reported in 2010 the synthesis of an amino acid employed as a tracer in chemotherapy via a Bucherer-Bergs-Strecker reaction as a key step [80]. This tandem-type methodology began with the disubstituted cyclopentene 418. Thus, ketone 419 was treated with KCN, ammonium carbonate, and ammonium chloride to obtain the product 420. After hydrolysis of hydantoin 420 to furnish compound 421 and several further transformations from this latter, precursor 422 was 18F labeled in 39 % radiochemical yield (Scheme 60). In this 3CR, majoritarian product 420 was found to be in a syn/anti (ca. 8:1) stereoselectivity. It was not possible to carry out the direct halogenation in the C-2 position concerning the spiro center in compound 420.
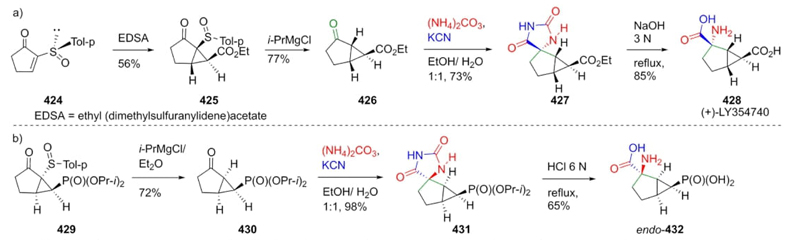
Midura, Mikołajczyk et al. developed a stereocontrolled strategy to prepare compound 428 and its analogue 432 utilizing a Bucherer-Bergs 3CR [81]. Thus, the synthesis of 428 (LY354740) started with the treatment of sulfoxide 424 with ethyl (dimethylsulfuranylidene)acetate (EDSA) obtaining a mixture of diastereomers ent/exo 425. Cyclopentanone 426 was then obtained with by treatment of alcohol 425 with isopropyl magnesium chloride. Then, oxobicyclo 426 was sequentially combined with potassium cyanide and ammonium carbonate to afford the spiro-compound 427. Finally, the synthesis of biomarker 428 was completed through hydrolyzation of its precursor 427 in 26 % overall yield from the sulfoxide 424. To obtain the (+)-LY354740 analogue 432, it was used the same strategy but needing only three experimental steps. From phosphonate 429, sulfur moiety was removed and promoted by isopropyl magnesium chloride to give the bicyclo 430. Thus, applying Bucherer-Bergs conditions to the later compound, the precursor 431 was assembled. Finally, its hydrolysis afforded the product endo-432 in 45 % yields by the three-steps (Scheme 61). In both strategies, a single diastereomer was afforded, where Bucherer-Bergs reaction was the key step.
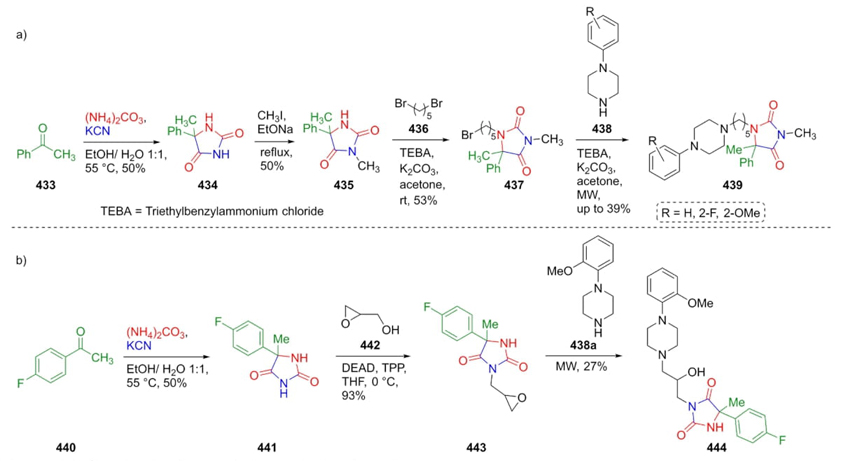
In 2014, Handzlik et al. used a Bucherer-Bergs reaction as the key experimental step to achieve a series of arylpiperazine-containing compounds which were evaluated as 5-Ht7 receptor ligands [82]. There are two synthetic procedures. The first one began with the combination of acetophenone (433), KCN, and ammonium carbonate to assemble the corresponding hydantoin 434 in 50 % yield, followed sequentially by an alkylation in different basic conditions to give the compound 435. Then, after a nucleophilic substitution with dibromide 436, the compound 437 was obtained. After a second nucleophilic substitution of this latter with piperazines 438, the spaced-type bis-heterocycles 439 were obtained. The second procedure involved also in a Bucherer-Bergs reaction starting from 4-fluoroacetophenone (440) to furnish hydantoin 441. After the one-pot process, compound 444 was formed through a Mitsunobu reaction coupled with an N-alkylation under microwave conditions (Scheme 62). According to the pharmacological studies carried out in this work, the existence of a substituent in position 3 of the hydantoins favors the ligand-protein affinity. In this strategy, two modifications were made in positions 1 and 3 of the hydantoin moiety, being the most favorable in position 3 by incorporating lipophilic groups, potentiating their affinity to the 5-Ht7 receptor ligand, which is involved in neurodegenerative disorders like the Alzheimer disease.
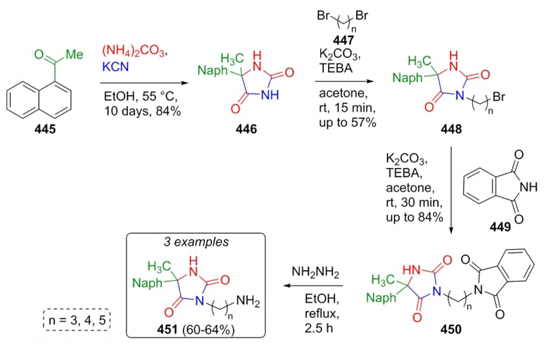
Later in 2015, Handzlik et al. again reported a procedure to achieve some new imidazolidine-4-ones via a Bucherer-Bergs as the key reaction of the process. The products were evaluated as chemosensitizers of S. aureus [83]. The naphthylacetophenone (445) was subjected to a Bergs-Bucherer reaction promoted by KCN and ammonium carbonate to afford the imidazolidine-2,4-dione (446). After two sequential C3-C5 alkylations (the first one on hydantoin 446, and the second one in compounds 448), phthalimides 450 were treated with hydrazine to finally afford products 451 (Scheme 63). Further modifications in N-3 position on the hydantoin ring are expected to potentiate the biological activity of such compounds.
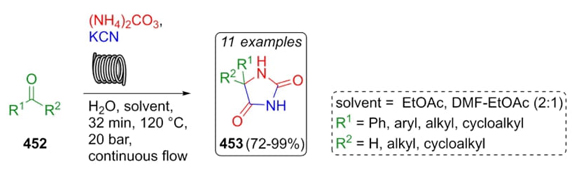
Kappe et al. reported the development of a creative and interesting approach to synthesize hydantoins via Bucherer-Bergs reaction under continuous flow conditions [84]. It was performed a two-feed setup with an organic solution of carbonylic compounds 452 and an aqueous solution of KCN and ammonium carbonate to afford hydantoins 453 in moderate to excellent yields (72-99 %) (Scheme 64). By employing polar solvents like alcohols, an unexpected issue of clogging occurs, which was easily solved by increasing the temperature of the system. However, in the case of alcohols, there is not a correct mixture of phases, so it was necessary to employ ethyl acetate or a mixture of solvents such as DMF-EtOAc in 2/1 v/v proportion.
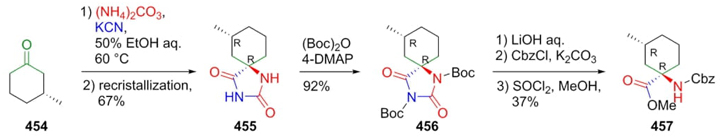
Tanaka et al. developed a new protocol to prepare peptides via the Bucherer-Bergs reaction used to increase annulated amino acids-chains [85]. The first step is just a Bucherer-Bergs reaction starting from chiral ketone 454 to give hydantoin cyclic-derivatives 455 in diastereomeric major product with R, R configuration (Scheme 65). After a separation of diastereomers, pure spiro-compound 455 was N-protected with Boc to give the product 456. Then, it was hydrolyzed with LiOH, and the carboxylate esterified to give the product 457. Stereoselective Bucherer-Bergs reaction is carried out due to steric hindrance between 1,3-diaxial H and C=NH groups. Moreover, it is worth noting that the cyclohexane ring has an axial methyl making a repulsion with to amino esther type group in 457.
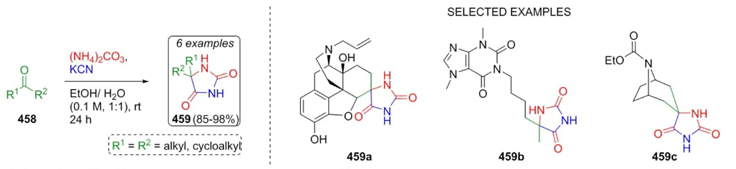
In 2016, Tomohara, Adachi et al. worked on the synthesis of hydantoins from extracts of Curcuma zedoaria via a Bucherer-Bergs reaction [86]. This strategy involved a direct chemical derivatization to synthesize the hydantoins 459. Thus, ketones 458 were combined sequentially with KCN, and ammonium carbonate to afford the products 459 in good to excellent yields (85-98 %) (Scheme 66). In the case of reactions with natural products, at high temperatures, it could produce undesirable compounds or fragmentation of the hydantoin ring in such compounds. On the other hand, it also could be produced other undesirable side reactions if concentrations of reagents are not optimal due to the wide variety of functional groups contained in such natural products.
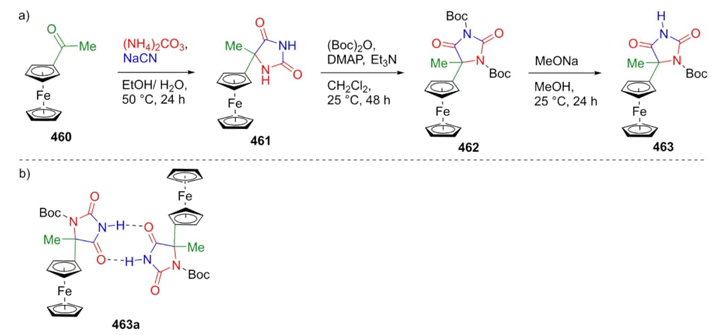
Santi et al. have been focused on the synthesis of aminoacids containing the ferrocenyl-moiety as a metallocene unit to provide H-bonded dimers through the Bucherer-Bergs reaction [87]. Thus, acetylferrocene (460) was subjected to mild Bucherer-Bergs conditions to give the product 461. Then, N-Boc protection was performed under the treatment of hydantoin 461 with (Boc)2O, DMAP, and triethylamine to give the bis-N-Boc-protected hydantoin 462. Finally, compound 462 was combined with sodium methoxide in methanol to afford the product 463 (Scheme 67(a)). Dimerization of hydantoin derivative 463 was carried out in a solution of CDCl3 (Scheme 67(b)). These kinds of compounds are suitable for recognizing cationic species due to the strategic position of the ferrocene fragment.
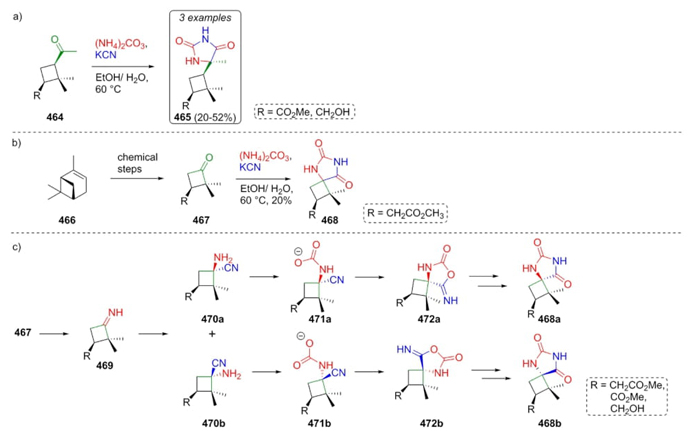
In 2018, Moglioni et al. reported the synthesis via Bucherer-Bergs of a series of some new hydantoins, which contain cyclobutyl rings [88]. Cyclobutanones 464 and 467 were sequentially combined with KCN and ammonium carbonate in water-methanol as the solvent system at 60 °C affording two types of hydantoins 465 and 468 (Scheme 68(a),(b)). A plausible mechanism for the synthesis of hydantoins 468 was displayed in Scheme 68c, where the main feature involves a carbamate-type intermediates 471a and 471b, which determine diastereoselectivity since they do not form diastereomeric mixtures in the conversion of precursors 467 into products 468, producing only a compound isolable while in the process from precursors 464 into 465 achieved a mixture of products.
Ritter-type 3CRs
In 1948, John J. Ritter developed a pioneering method to synthesize amides. Formation of carbocations 475 coming from alkenes 473 or alcohols 474 in an acid medium is the first step of this methodology. Then, addition of nitriles 476 furnished nitrilium ion intermediates 477 and their resonance form 478, which in presence of water (or other kind of nucleophiles) finally provide the amides 479, eventually in good to excellent yields (Scheme 69) [89]. Even, Ritter reaction has been used in an intramolecular way to give heterocycles, for instance, lactams and other kind of related aza-heterocycles [90].
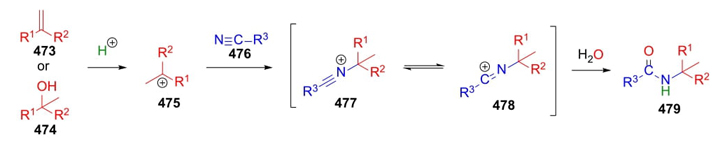
In 2011, Togni et al. developed a Ritter-type reaction to generate compounds with N-CF3 bonds [91]. In this approach, triazoles 480 were combined sequentially with iodine-containing compound 481 in acetonitrile under mild acidic conditions to furnish N-(trifluoromethyl)imidoyls 483 as the main products in modest yields (37-63 %) (Scheme 70(a)). It was proposed a plausible catalytic mechanism, where protonated iodine-containing compound 481 and acetonitrile 482 gave the complex 485. Then, nitrilium ion 487 was formed through a reductive-elimination step. Imines 483 were constructed by a reaction between triazoles 480 and ion 487. Finally, it was released a proton to complete the catalytic cycle (Scheme 70(b)). This strategy allows the formation of a N-CF3 bond since regularly it was formed through another kind of methodologies. Two by-products were formed due to stereoelectronic nature of reactants. For instance, an excess of triazole 480 promotes a trifluoromethylation process, while an excess of iodaoxole (481) produces a Ritter addition of nitrile, which was controlled by stoichiometric quantities of the corresponding reactants 480 and 481.
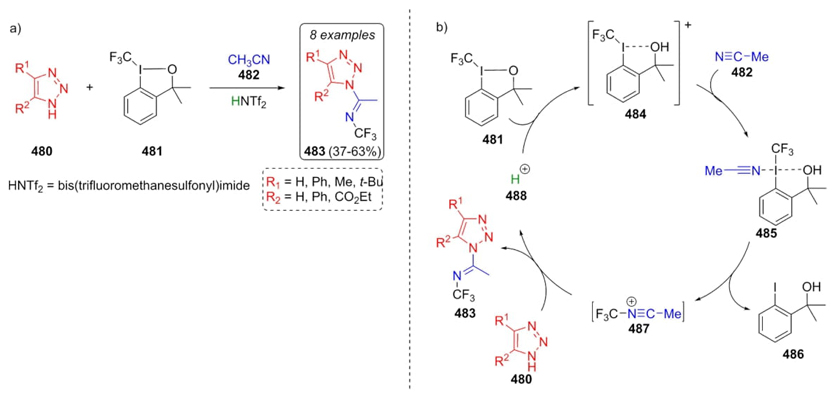
Johnston et al. reported in 2011 the total synthesis of Hapalindoles K, A, and G in twelve steps where a Ritter reaction was the key one [92]. Starting reagents 489 and 490 were previously synthesized. Thus, it was performed a Diels-Alder cycloaddition between them (489 and 490) to give the compound 491, which was reduced with DIBAL to provide alkene 492. This latter was treated with TBAF to obtain the alcohol 493. Then, reagents 492 or 493 were subjected to acidic Ritter conditions with TMSCN affording the compound 494 in good stereoselectivity. After several chemical steps, the synthesis of hapalindole K was completed (Scheme 71). It is noteworthy that Ritter reaction was useful for stereoselective formation of a C-N bond.
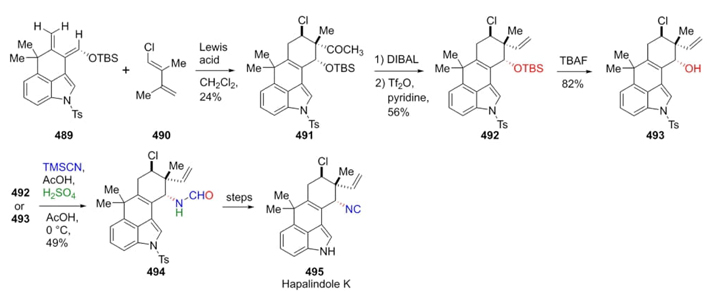
In 2012, Lavilla et al. developed a unusual multicomponent approach through a tandem Mannich-Ritter-type strategy [93]. Thus, unsubstituted compounds 496 and imines 497 were sequentially combined with nitriles 499 to afford products 501 in yields up to 69 % under Lewis-acid catalytic conditions at room temperature. Plausible mechanism comprises the formation of Mannich-type intermediates 498 from olefine and imine reagents, followed by the addition of nitriles to give the Ritter-intermediates 500. Finally, amidines 501 were furnished by means of an intramolecular C-N bond formation (Scheme 72). As first step, it was proposed that cyclic compounds 496 and imines 497 react via Povarov reaction but, it was observed a high ring strain in that intermediate formed so that the mechanism pathway was directed to run via a Mannich sequence. This reaction works well with some restrictions: a) it was necessary to carry out the reaction using an excess of nitriles; b) some compounds such as dimethylindolenine do not perform MCR due to steric hindrance; c) other products with poor yields were the aromatic imine derivatives which were epimerized; d) it was used different N-olefins such as lactams, enamides or thiazolones, which afforded Mannich-type products.
Yadav et al. reported in 2013 a creative three-component procedure involving a cascade Prins-Ritter process as a key step to synthesize tetrahydropyranes as precursors of anti 1,3-aminoalcohols [94]. To demonstrate the effectiveness of this approach, it was achieved the total synthesis of (-)-Halosaline. Chiral alcohol 503 was previously prepared starting from epoxides through Jacobsen’s hydrolytic kinetic resolution followed by a protection. Thus, aldehyde 502, alcohol 503, and nitrile 504 were sequentially combined using a Lewis-acid as catalyst to give product 505, which was treated with sodium iodide to give acrylamide 506. Eight steps from compound 506 were performed, some of them were a ring closing and alkene reduction to complete the total synthesis of (-)-Halosaline (507) (Scheme 73). After MCR, substituents found in equatorial position allowed stereocontrol behind the synthesis of anti-1,3-aminoalcohols, which can be further functionalized.
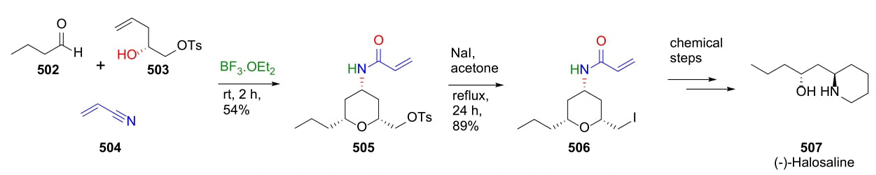
Scheme 73 Synthesis of anti 1,3-aminoalcohols via a strategy involving a Prins-Ritter cascade sequence.
In 2013, Reddy et al. completed and reported for the first time the total synthesis of the alkaloid (+)-8-ethylnorlobelol (511) via a multistep strategy in which a Prins-Ritter cascade reaction was the key process [95]. Synthetic procedure began with the chemoselective protection of dialcohol 508 with tosyl group selectively in the primary alcohol affording product 509. Combination of secondary alcohol 509 with n-propanaldehyde and acrylonitrile under Lewis acid catalysis resulted in the formation of cis-diastereomer of tetrahydropyran 510. Total synthesis was completed in ten steps with high diastereoselectivity (Scheme 74). In the same way, the stereoselectivity key feature was directed since the cascade multicomponent process.

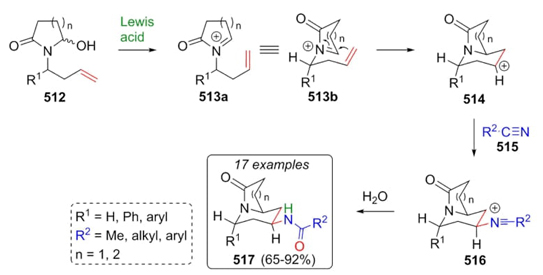
Saikia et al. developed an interesting tandem sequence via an aza-Prins reaction coupled with an intermolecular Ritter reaction to give azabicyclic species 517 [96]. Previously, it was prepared starting reagents 512. Thus, lactams 512 were mixed with nitriles 515 under Lewis acid catalysis to achieve azabicyclic products 517 in good to excellent yields (65-92 %) and with high diastereoselectivity. It was suggested a plausible mechanism where iminium ions 513 were promoted by a Lewis acid. Then, carbocations 514 in chair-like favored conformation lead trapping nitriles 515 affording nitrilium ions 516, which were hydrolyzed completing the synthesis of products 517 (Scheme 75). It was observed that Lewis acids have a favorable effect on the yields and diastereoselectivity, better than Brønsted acids. Also, yields of this MCR depend directly on substituents from the allylic chain, giving the best yields to electron-withdrawing groups. Endo-trig cyclizations of compounds 513 were carried out in axial substitution due to steric hindrance between substituents R1 and carbonyl group from N-acyliminium ions as well as strong ring strain.
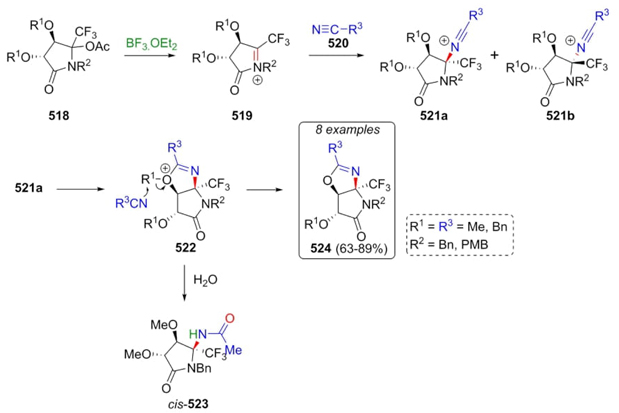
The synthesis of new pyrrolidin-2-ones reported by Grellepois, Ben-Jamaa et al. involved a diastereoselective Ritter-type reaction [97]. The first step was the formation of N-acyliminium ions 519, which were trapped by nitriles 520 affording isomeric mixtures of nitrilium ions 521a and 521b. Then, two routes continued from isomers 521a: 1) cis-nitrilium ions 521a promoted the synthesis of oxazolines 524 in 63 to 89 % yields through intermediates 522, and 2) hydrolysis of intermediates 522 allowed the formation of cis-amides 523 (Scheme 76). These MCRs were carried out under Lewis acid conditions. During the processes, side products like amides or a diastereomeric mixture of products were obtained due to the presence of water in reaction media, which was solved using adequate quantities of acetonitrile to obtain 521a as the major products. In contrast, compounds 521b were obtained as by-products.
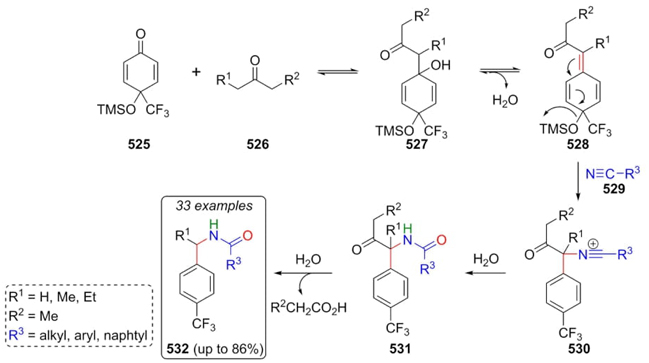
Li, Liu et al. developed an interesting method for preparing N-benzyl amides 532 through a cascade Ritter-type reaction [98]. In this process, the enolic form of ketones 526 performed a nucleophilic attack on carbonyl compounds 525 generating the intermediates 527. Dehydration of these later affords intermediates 528, which were trapped by nitriles 529 gave the nitrilium ions 530. Thus, after their hydration to give compounds 531, the elimination of leaving group completed the synthesis of p-(trifluoromethyl)benzyl amides 532 in up to 86 % yields (Scheme 77). As seen, best yields were obtained using dichloroethane as solvent, and TMSCl as Lewis acid catalyst, and water as an additive. In particular, the addition of water promoted hydrolysis of acyl group. Regarding to nitriles, multisubstituted benzonitriles do not promote reaction due to steric hindrance. On the other hand, benzonitriles substituted in ortho or para position decrease considerably the yields due to electronic inductive effects. A variety of electron-donating and electron-deficient groups were used in this approach, which indicates the viability of this process.
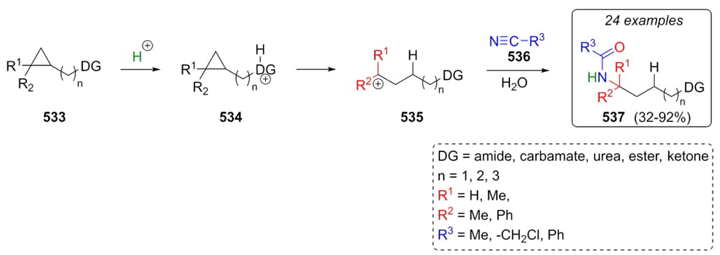
Protonolysis of cyclopropane-containing compounds was developed by Jirgensons et al. in 2019. This strategy makes use of amides, carbamates, ureas, esters, and ketones as reagents to generate corresponding amides via a Ritter-type reaction [99]. Thus, cyclopropanes 533 were protonated in a directing group (amide, carbamate, urea, and so on) through proton C-C transference to form the carbocations 534. Products 537 were achieved through cations 535 trapping the nitriles 536 (Scheme 78). The reaction was carried out in acidic media under mild conditions. The use of trifluoroacetic acid, chloroacetonitrile, and acetic acid in a 1:1:1 proportions and heating the reaction at 60 °C gave the best yields. When a phenyl substituent was placed into the cyclopropane ring, the yields decreased considerably due to its interference at protonolysis step. In the same way, phthalimide and urea as directing groups of the reaction afforded the products in low yields, obtaining various byproducts.
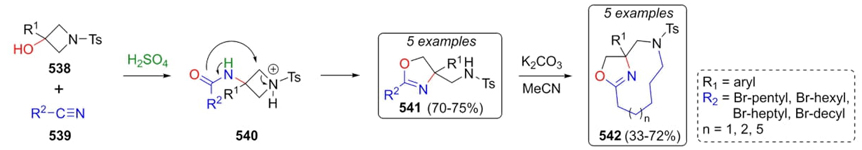
An elegant methodology developed by Baxendale et al. towards the synthesis of the new and complex macrocycles 542 involved a Ritter-type reaction as the first step of such procedure [100]. To achieve oxazolidines 541, Ritter conditions from alcohols 538, nitriles 539, and sulfuric acid afforded amides 540, where the oxygen of the amides attacked the ring releasing the ring strain to give the products 541. With 4,5-dihydrooxazoles 541 in hands, compounds 542 were synthesized via intramolecular cyclizations in basic conditions (Scheme 79). The corresponding Ritter-intermediates were not formed in this approach. Moreover, in this strategy, several acids were evaluated, sulfuric and fluoroboric acids were the best ones, where sulfuric acid was chosen for its economy and ease of access. On the other hand, authors examined solvent effects in chloroform, dichloromethane, toluene, xylene, among others; DCM was selected as the best one.
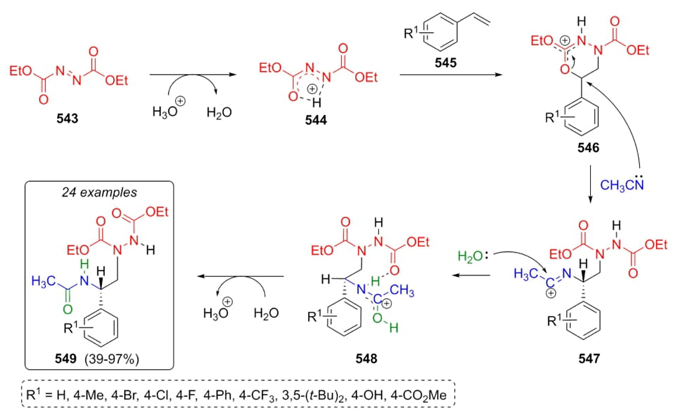
Bao et al. reported in 2021 the synthesis of vicinal diamines 549 via a Ritter-type multicomponent reaction [101]. The optimal reaction conditions were using the strong Brønsted acid HPF6, and Cu(CH3CN)4PF6 as catalyst . Diethyl azodicarboxylate (543) and styrenes 545 were mixed to achieve the corresponding products in moderate to good yields (39-97 %). Authors proposed a plausible reaction mechanism, that was supported by DFT calculations. Thus, azodicarboxylate (543) was protonated by a Brønsted acid to assemble the intermediate 544, which in the presence of styrenes 545 afforded carbocations 546. Finally, products 549 were obtained following a Ritter reaction route after a nucleophilic attack of acetonitrile to intermediates 546, and the addition of water to carbocations 547 and dehydration of intermediates 548 (Scheme 80). Acetonitrile not only worked as the solvent but also contributed to the multicomponent process as one of the three reagents. Electronic effects influenced the yields because in the substitution from compounds with electro-donating or electro-withdrawing groups, the ortho or meta positions in the aromatic moiety gave the best yields, whereas in the para position, the yields were lower. In addition, the yields are affected by the amount of Brønsted acid used.
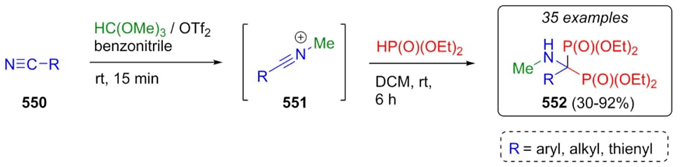
In 2022, Ye, Huang et al. worked on the synthesis of bisphosphonates through an interrupted version of Ritter reaction [102]. The methodology started with nitriles 550, and their sequential combination with trimethyl orthoformate and triflic anhydride (1:1), nitrilium ions 551 were afforded. Then, These later ones were trapped by phosphite ester to assemble the bisphosphonates 552 in moderate to good yields (30-92%) (Scheme 81). Yields were affected by electronic effects of electron-withdrawing groups and steric hindrance in bulky groups from benzonitriles such as 4-tert-butyl or 4-propylcyclohexyl ones, as well as ortho-methyl-substituted benzonitriles and para-halogenated-substituted benzonitriles. Instead that, alkylic nitriles gave the best yields.
Kabachnik-Fields-type 3CRs
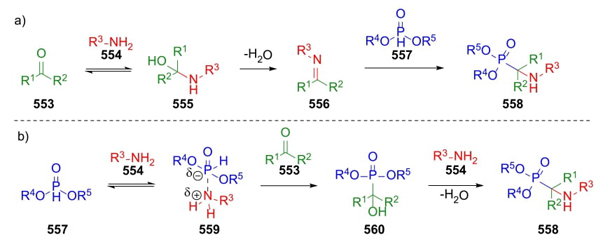
Another multicomponent reaction was developed in 1952 by Martin I. Kabachnik and Ellis K. Fields. This 3CR involves a reaction between carbonylic compounds (usually aldehyde but, a few examples using ketones have been reported), an amine, and a dialkyl phosphite [103]. The mechanism may take two pathways depending on the nature of reagents and mainly of the order of addition: a) condensation between carbonyl compounds 553 and amines 554, followed by addition of P-H anions 557 to the corresponding imines 556 to complete the synthesis of phosphonates 558; or b) addition of phosphites 557 to amines 554 to afford phosphonate complexes 559. Then, zwitterions 559 are mixed with carbonyl compounds 553 to achieve compounds 560, which through a condensation with amines 554 furnish products 558 (Scheme 82(a),(b)) [104].
Ordóñez et al. developed a synthetic methodology based on the Kabachnik-Fields three-component reaction to synthesize a few isoindolin-1-one-3-phosphonates [105]. Thus, condensation between aldehydes 563 and amines 564 afforded iminium ions 565, which were nucleophilically attacked by phosphites 562. After a dehydration process, products 567 were obtained under solvent and catalyst-free conditions (Scheme 83). It must be noted that the determinant step that allowed the high degree of diastereoselectivity is the nucleophilic attack of phosphites to imines due to a less hindered attack in re-face.
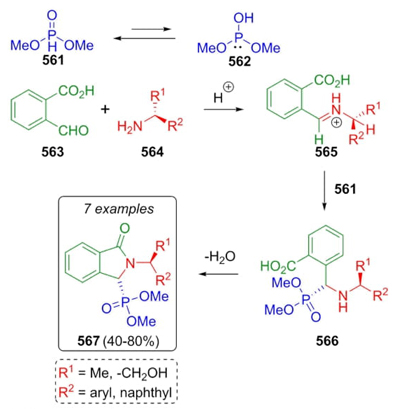
Berlicki et al.reported in 2012 the use of Kabachnik-Fields reaction to synthesize some new hydroxmethylphosphonic acid[106]. This synthetic procedure was used to evaluate the inhibitory properties of synthesized compounds towards bacterial urease. Thus, aldehydes 569 and phosphinic acid (568) were combined to achieve the a-hydroxy phosphinic acids 570, which were trapped with secondary amines 571 followed by addition of formaldehyde in acidic conditions to afford the bioactive compounds 572 in yields up to 81 %. Phosphinic acid derivatives 573 were obtained by further catalytic hydrogenation of compounds 572 (Scheme 84). This procedure involves a modification of the amine moiety and the incorporation of a hydroxymethyl fragment for modulating biological activity.
Seidel et al. worked on a new Kabachnik-Fields-type reaction for the preparation of α-amino phosphine oxides (Scheme 85) [107]. This synthetic strategy involves a sequential combination of aldehydes 574, cyclic secondary amines 575, and phosphine oxides 576 using benzoic acid as the catalyst and under microwave heating conditions to give the α-amino phosphine oxides 577 and 578 in moderate to good yields (up to 89 % in the sum of yields for both isomers) and regioselectivity 1:1. The procedure promoted formation of C-P bond through functionalization of C-H bond using electron-withdrawing aldehydes and carboxylic acid as catalyst which accelerate the iminium ion formation.
Čikotienė et al. reported the preparation of many condensed pyrrolylphosphonates through a Kabachnik−Fields reaction-based methodology [108]. With reagents in hand, they proceeded to implement Kabachnik−Fields process through a sequential combination of aldehydes 579 with anilines 580 and dimethylphosphite under copper (I) catalyst to give the complex intermediates 582, which performed an intramolecular cyclization to prepare a set of phosphonate-containing cyclized products 583 or 584 in moderate yields (48-69 %) (Scheme 86). Electron-releasing groups placed in aldehydes do not promote the reaction. On the other hand, pyridines and quinolines activate the triple bond of alkyne moiety, which decreases electron density from imine, thus promoting nucleophilic attack of phosphite via an endo-dig cyclization.
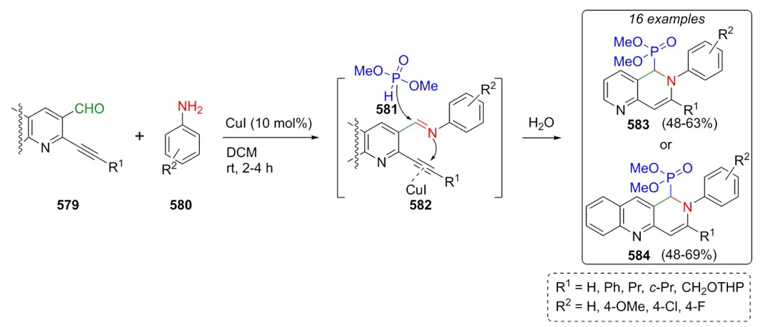
Based on recent studies of fluorescent nanoparticles, Wan, Zhang, Wei et al. developed the first synthetic procedure to prepare active organic nanoparticles via the Kabachnik-Fields reaction, which were further evaluated in HeLa cervical cancer cell line [109]. Aggregation-induced emission-active aldehyde 586 was mixed with polyetherimide 585 and diethyl phosphite (587) under solvent and catalyst-free conditions using ultrasound heating affording product 588 in moderate yield (Scheme 87). This compound was synthesized under environmentally friendly conditions, and it exhibits potential activity for bioimaging due to its amphiphilic character. Likewise, this polymer may work as a cell biomarker because it exhibits biocompatibility with HeLa cell line.
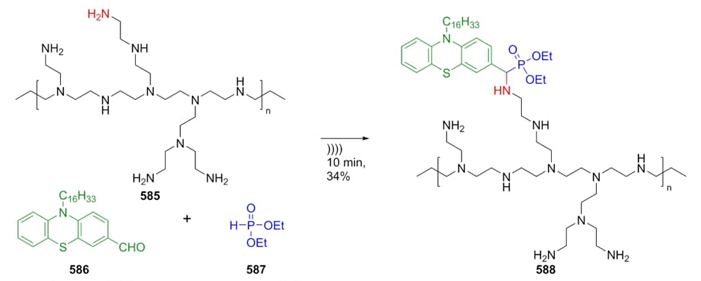
In 2018, Wilhelm, Silva et al. reported the synthesis of twelve novel selanyl-a-amino phosphonates via a Kabachnik-Fields reaction [110] (Scheme 88). After the previous preparation of selenide amines 590, the combination of aldehydes 589 with selenide amines 590, and phosphonates 592 under solvent-free conditions catalyzed by niobium oxide allowed the preparation of organylselanyl α-amino phosphonates 593 in moderate to good yields (48-90 %). The use of diphenylphosphite and 3,5-dimethoxybenzaldehyde decreased the yields. Instead that, aldehydes substituted at the ortho position gave the best yields in the same way that the aryl groups in the selenide amine. These compounds were evaluated behind their antioxidant properties, as well as enzymatic inhibition properties.
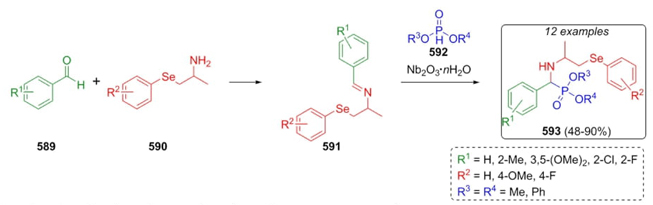
Nikalje et al. worked in 2018 on the synthesis of some new phosphonate-containing indolinones via a Kabachnik-Fields reaction. The synthesized compounds were probed against a panel of cancer cell lines such as MCF-7, IMR-32, SK-MEL-2, MG-63, to name a few [111]. Starting from indoline 594, it was was formed the hydrazone 595 in the presence of hydrazine and acidic conditions. Thus, aldehydes 596, hydrazone 595, and triethylphosphite (598) were sequentially combined in the presence of ceric(IV) ammonium nitrate as a green catalyst at room temperature to synthesize the products 600 in good to excellent yields (70-95 %) (Scheme 89). Mechanism suggests that first were achieved imines 597. Then, the catalyst promotes imines 599 formation, as well as the nucleophilic attack of phosphite 598 to give the intermediates 599. Finally, water in the media reacts with phosphonium ions to achieve the Kabachnik-Fields products 600. It is noteworthy the environmentally friendly conditions (EtOH, AcOH and ultrasound irradiation) involved in this synthetic strategy as well as potential biological activity on cancer cell lines.
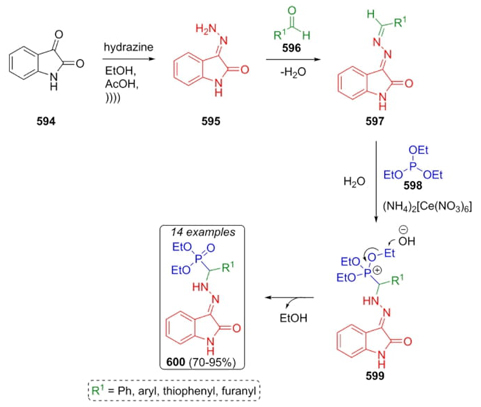
In 2019, Bálint et al. developed a methodology consisting of a double Kabachnik-Fields from aminoalcohols as key reagents (Scheme 90) [112]. Thus, paraformaldehyde (601), 2-aminoethanol (602), and phosphine oxides 603 were reacted together using microwave irradiation as a heat source in acetonitrile as a solvent and under catalyst-free conditions to afford products 604 and 605 in good yields (up to quantitative yields in the case of 605). In this procedure, it was a functional range of temperatures (from 80 to 100 °C), and a quantity of reagents that gave both products. Authors found also that the use of an excess of paraformaldehyde at 80 °C, promotes the formation of both Kabachnik-Fields products.
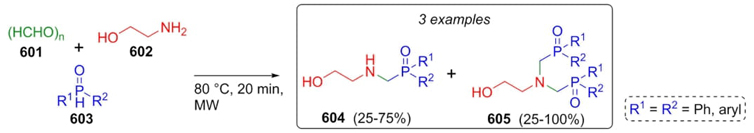
Ordóñez et al. reported in 2019 a new strategy to synthesize 1,2,3,4-tetrahydroisoquinoline-1-phosphonic- and -1-phosphinic acids 610 via a Kabachnik-Fields reaction (Scheme 91) [113]. Thus, aldehyde 606, amines 607, and phosphites 608 were combined in acetonitrile at 60 to 85 °C under catalytic amounts of phenylboronic acid (609) (10 % mol) to afford the products 610 in moderate yields (70-75 %). This study aimed to obtain analogues of 1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid through isosteric replacement of phosphonic acid instead of carboxylic acid fragment via an MCR-based synthesis under mild reaction conditions.
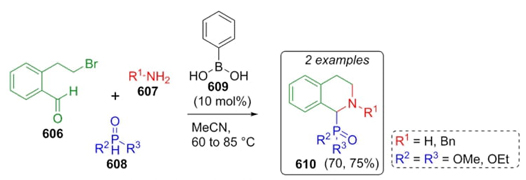
Bazine et al. developed a rapid and efficient methodology to prepare new α-amino phosphonates through a Kabachnik-Fields reaction as a key step. The synthesized compounds were evaluated as antioxidants and enzymatic inhibitors (Scheme 92) [114]. To prepare the quinoline-type reagents 612 it was employed a Meth-Cohn multistep reaction sequence starting from amides 611. Then, compounds 612 were treated under mild acidic conditions to afford oxoquinolines 614. Thus, quinolines 614, amines 615, and triethyl phosphate (618) were then subjected to Kabachnik-Fields reaction conditions in liquid ionic TEAA (triethylammonium acetate) using ultrasound irradiation to synthesize the corresponding products 613 and 617 in moderate to good yields (up to 93 %). Liquid ionic TEAA promoted high yields and in short reaction time. In addition, this procedure has a few advantages over previous ones. For instance, short reaction times, high yields in one-pot procedure, the products were afforded without purification by chromatography, and ease of experimental operation.
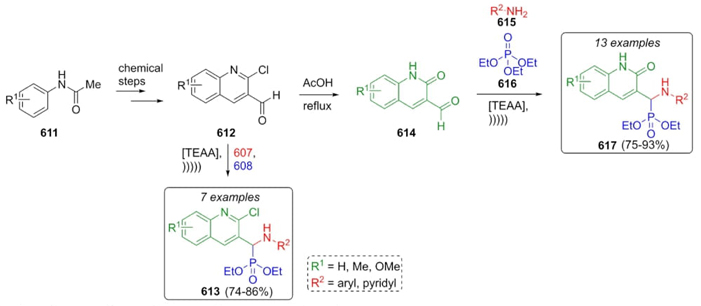
Guezane-Lakoud et al. developed a synthetic strategy towards the synthesis of new phosphates a-aminophosphonates through a Kabachnik-Fields-3CR as the first step of such procedure, followed by an Atherton-Todd reaction [115]. Sequential combination of hydroxybenzaldehydes 618, anilines 619, and diethylphosphite (620) using nickel(II) sulfate as a catalyst allowed the formation of the new α-aminophosphonates 621 in moderate to good yields (65-92 %) (Scheme 93). Then, phosphates α-aminophosphonates 622 were obtained through an Atherton-Todd reaction condition from compounds 621. Precursors with substitution at ortho and para positions of benzaldehydes afforded the best yields in comparison to those from benzaldehydes substituted in meta position. The use of anilines with electron-rich or deficient groups did not affect the yields. Also, it is worth highlighting the solventless conditions, room temperature, and short times for all the processes.
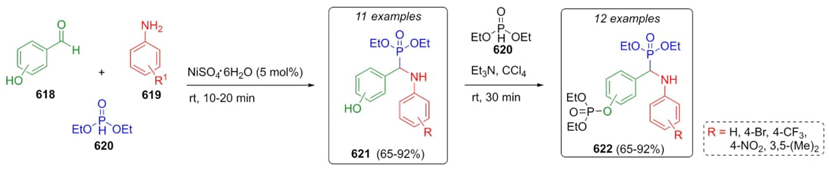
Hernández-Fernández, Robledo-Leal, López-Cortina et al. reported the synthesis of a-aminophosphonates and their antifungal evaluation against L. prolificans [116]. Thus, aromatic, and heterocyclic aldehydes 623, para-substituted anilines 624, and diphenyl phosphite (625) were reacted together in a one-pot process using MW as a heat source without catalysts to produce the a-aminophosphonates 626 in moderate to good yields (11-85 %) (Scheme 94). It is important to highlight the eco-friendly reaction conditions that were used. Low reactivity was observed in some products probably due to the use of low activated 4-dimethylaminocinnamaldehyde, which promoted the formation of undesirable side products.
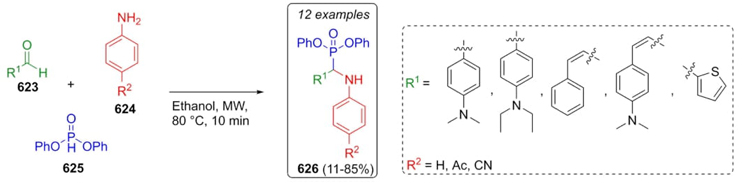
Gewald -type 3CRs
The reaction developed by Karl Gewald in 1966 has two experimental ways to be carried out. One of them is a 3-CR [117]: a) the first version (Gewald-2CR): mercaptocarbonyl reagents 628 are condensed with base-activated nitriles 627 to achieve intermediate-type mercaptonitriles 629, which after a rearrangement/cyclization process on compounds 630, 631 and 632 gave the 2-aminothiophenes 633 (Scheme 95a); and b) second version (Gewald-3CR): the first step consists of a Knoevenagel condensation between carbonyl compounds (aldehyde or ketone) 635 and base-activated nitriles 634 to give cyano alcohols 636, that after a deprotonation achieved intermediates 637, and a dehydration to furnish intermediates 638 was ensembled towards the corresponding acrylonitriles 639, which were thiolated with elemental sulfur at active methylene position to give the sulfur-containing-intermediates 640. Finally, 2-aminothiophenes 633 were furnished through annulation promoted by intramolecular nucleophilic attack by mercaptide in precursors 641 (Scheme 95b) [118].
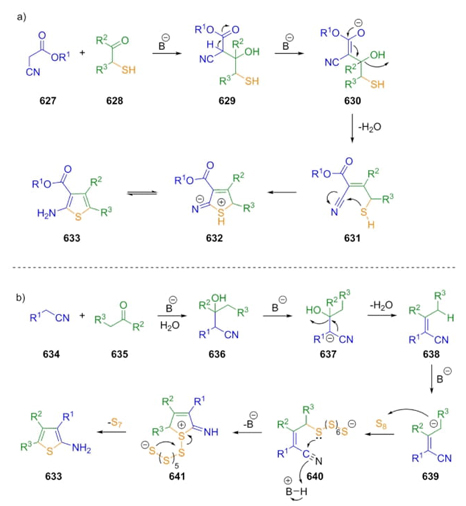
Reissig et al. developed a synthetic procedure to synthesize complex macrocyclic peptidomimetics containing the 5-aminothiophene moiety as the main building block [119]. For this one, it was necessary to improve a Gewald three-component reaction. Thus, after combining cyclopropanecarboxylate 642, methylene-activated nitriles 643, and elemental sulfur, thiophene 644 was afforded in a sequence involving a ring-opening process from compound 642 and a further condensation with nitrile 643 and elemental sulfur (Scheme 96). Amine functionalization of compound 644 furnished the product 645, which was used as unit for construction of the peptidic macrocycle 646. The key step in this methodology was the Gewald reaction, which allowed the incorporation of thiophene fragment into macrocycle 646.
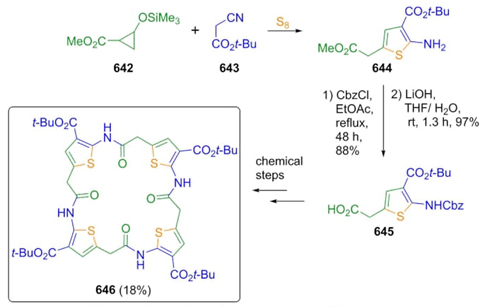
In 2011, Skene et al. synthesized a couple of fluorescent aminobisthiophenes via a Gewald reaction as second step of both synthetic procedures (Scheme 97) [120]. Starting from 2-thiophene carboxaldehyde (647), trimethylsulfonium iodide and SiO2 were added to give the 2-thienylacetaldehyde (648), which was combined under a Gewald 3CR protocol with nitrile-ester 649 and sulfur to obtain the product 650 in 25 % yield per two steps. Compound 650 was then formylated using POCl3 under Vilsmeier-Haack conditions. Then, Base bis-thiophene containing Schiff 652 was prepared by treatment of aldehyde 647 with bis-thiophene 651. Additionally, subsequent reductive alkylation of aminobisthiophene 651 with benzaldehyde (653) achieved the bis-thiophene-containing product 654. Gewald reaction allowed incorporating a second thiophene ring, which increases the optical activity of the whole compounds helped by the presence at the same time of electron-rich and deficient groups (push-pull effect).
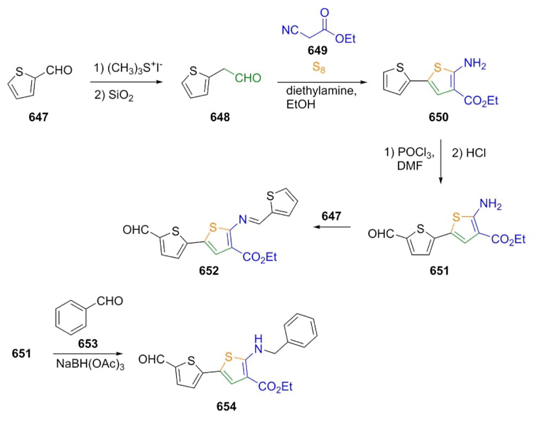
Hesse et al. reported a rapid and efficient procedure to prepare the new 5-aryl 2-aminothiophenes 657 via a Gewald reaction under microwave heating conditions (Scheme 98) [121]. Thus, aldehydes 655, nitriles 656, and elemental sulfur were mixed under microwave- heating conditions to achieve the products 657 in moderate to good yields (50-99 %). Also, the synthesis of 657 was performed but under conventional heating conditions, and the products were afforded in lower yields. It can be noted that microwaves as a heat source are optimal for decreasing reaction times. This approach allowed synthesizing 2,4,5-substituted thiophenes in short times and with high chemical yields.
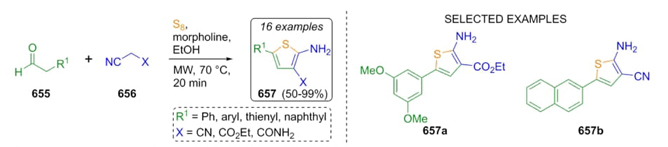
Based on the idea that polysiloxanes are environmentally friend catalysts, Lai, Xu et al. worked in 2012 on a creative and green approach, in which amine-functionalyzed polysiloxanes were used as catalysts in a Gewald reaction (Scheme 99) [122]. Ketones 658, methylene-activated nitrile 659, and elemental sulfur were sequentially mixed using the amino catalyst 660 to generate the aminothiophenes 661 in moderate to good yields (32-89 %). Ketones worked well in such reaction, regardless of low reactivity mainly by stereoelectronic effects. Thus, to solve the problem that ketones afforded low yields due to their limited reactivity, catalyst 660 helped the process successfully. The size of the ketone ring influenced yields, 6-membered ring ketones afforded the best yields instead of 5 or 7 membered ketones.
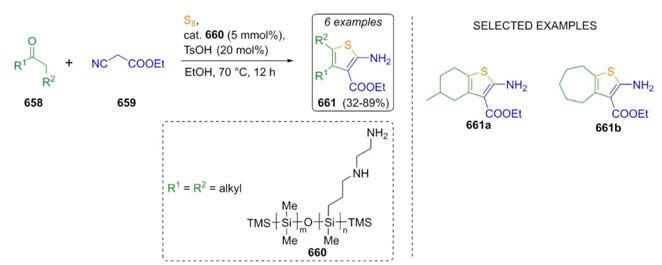
Pal et al. developed a methodology to synthesize anticancer agents containing the fused-type bis-heterocyclic thieno[3,2-c]pyran-4-one core [123]. Thus, the synthesis of the 2-aminothiophenes 664 as key reagents were carried out via a Gewald reaction through a sequential combination of cyclic ketones 662, ethyl cyanoacetate (663), and elemental sulfur. Further transformations involved Sandmeyer iodination to give compounds 665, cyclization promoted by iodide, and various C-C couplings catalyzed by palladium-based reagents from alkynes 666 achieved the compounds 667, and the products 668 (Scheme 100). The thiophene ring is the key fragment in this strategy because it was found in several biological active compounds. Gewald reaction allows often incorporating thiophene fragments in short reaction times and high yields.
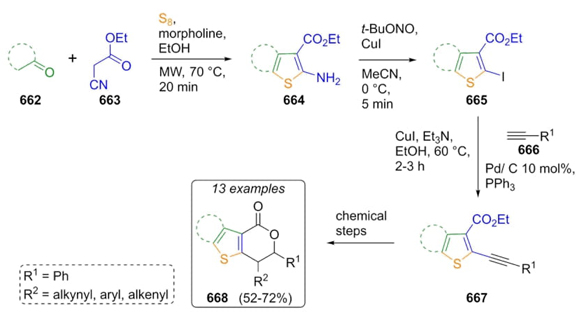
Parsa, Pal et al. developed a multistep methodology to prepare a series of fused-type thieno[2,3-d]pyrimidines 674, that include a Gewald 3CR as the first and key step. Synthesized compounds were evaluated as inhibitors of PDE4 enzyme [124]. Thus, cyclic ketones 669, ethyl cyanoacetate (670), and elemental sulfur were combined in morpholine to afford the products 671, which performed a ring closing promoted by formamide or formimidine acetate to give the products 672. Besides, compounds 673 were constructed by treatment of polyheterocycles 672 with stoichiometric amounts of POCl3. After a few further chemical steps, it was accomplished the synthesis of the target compounds 674 (Scheme 101). As seen, Gewald reaction acts as the key step in constructing thieno[2,3-d]pyrimidine core, which is present in a relatively wide variety of bioactive compounds, even in some current drugs.
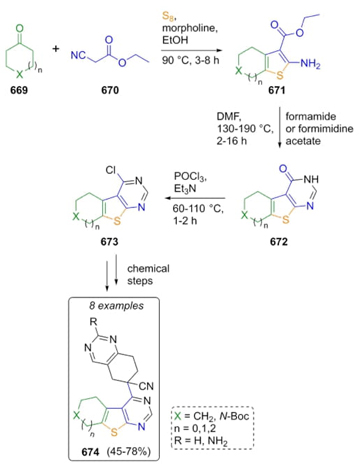
Kanizsai et al. developed in 2019 a synthetic strategy to obtain novel β-aminonitriles, for instance the 4,5,6,7-tetrahydrothieno[2,3-c]pyridines 677 and 679 via a Gewald reaction [125]. These compounds were evaluated as antitumoral by tyrosine-kinase inhibition. Thus, piperidones 675, malononitrile (676), and elemental sulfur were mixed under basic conditions to assemble the compounds 677 in moderate to good yields (10-87 %) (Scheme 102). Also, a few compounds 677 were hydrolyzed toward the formation of their amide-analogues 679 in moderate yields (15-59 %). A direct way to synthesize compounds 679 using a Gewald reaction was by combining the piperidones 675, cyanoacetamide (678), and elemental sulfur. This last pathway is less efficient than the procedure in two steps. In other way, products 677 and 679 have a potential antitumoral activity due to influence of N-sulfonyl pharmacophoric fragment.
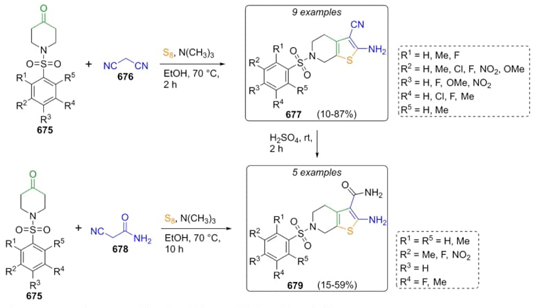
Nguyen, Mac et al. worked in 2021 on the synthesis of 2-aminothiophenes through a Gewald reaction [126]. Thus, α-cyanoacetates 681, chalcones 680, and elemental sulfur were combined under basic catalytic conditions to provide the 2-aminothiophenes 688 in moderate to good yields (48-86 %). Authors proposed a plausible mechanism where the first step was a nucleophilic attack of cyanoacetates 681 to chalcones 680 producing the anions 682 (Scheme 103). Steric hindrance inhibits Knoevenagel condensation as the first step. Then, DABCO-sulfur complex was attacked by anions 682 affording the products 683, which were cyclized to give the tetrahydrothiophenes 684 and came into equilibrium with dihydrothiophenes 685. Finally, an aromatization process occurs from compounds 686 and then 687 due to the presence of elemental sulfur to produce compounds 688. The sulphuration process is favored in the cis intermediate due to steric hindrance. The regioselectivity was promoted by appropriate chalcones. Moreover, there are no reports about the use of cyanoacetates and chalcones as starting materials in Gewald-type processes.
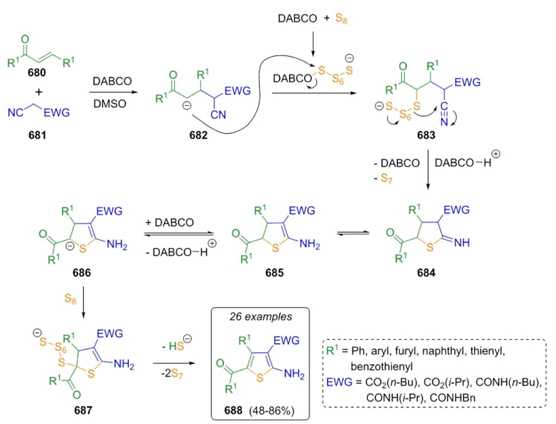
Yeh, Tsai, Lee et al. reported the synthesis of many thiophenes via a Gewald reaction, as well as the evaluation of synthesized compounds as Cisd2 (CDGSH iron sulfur domain 2) protein activators that cause childhood diabetes. [127]. A sequential combination of methylene-activated amides 690, nitriles 689, and elemental sulfur in mild reaction conditions achieved the new thiophene derivatives 691 in moderate to good yields (8-82 %) (Scheme 104). Synthesis of compounds was focused on the effect of substituents R1 or R2 on the activator Cisd2 to potentiate their activity. In the case of substituents R1, bulky esters decrease the activity as well as groups like amide or nitrile. On the other way, R2 substituents exhibited higher activity in 3 and 4-substituted phenyl rings with electron-donating groups such as methyl or methoxy, instead electron withdrawing groups that showed lower activity.
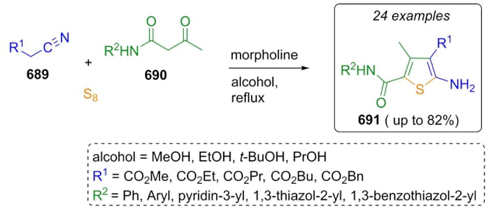
Petasis-type 3CRs
The Petasis reaction, also known as Petasis-Boron-Mannich reaction, was reported in 1993 by Nicos A. Petasis, who modified the original Mannich reaction by employing a boronic acid as a nucleophile. This three-component reaction involves a sequential combination of carbonyl derivatives with secondary amines and boronic acids. The reaction mechanism is still under discussion [128]. However, the most plausible and accepted one proceeds via a hemiaminal-type condensation between aldehydes or ketones 692 and secondary amines 693. Then, iminium ions 694 react with boronic acids 695 to give the complex 696, which through a C-C bond migration gave amine derivatives 697 (Scheme 105) [129].
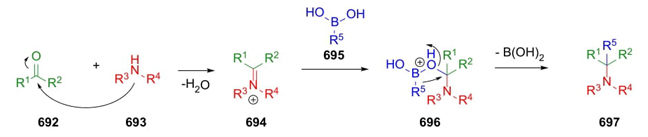
Pyne et al. reported in 2010 a procedure that allowed the total synthesis of Calystegine B4 using a Petasis reaction as the first key step [130]. Thus, a combination of (-)-D-lyxose (698) as a carbonylic compound, benzylamine (699), and boronic acid 700 afforded the product 701 in 82 % yield. Then, the amino group from compound 701 was protected using Boc to give the aminoalcohol 702, which was treated with trityl chloride to give the advanced intermediate 703. After seven additional transformations from this later, it was obtained the natural product 704 (Scheme 106). Notably, the Petasis reaction allowed obtaining stereoselectivity of the amino group, which could be used for further transformations.
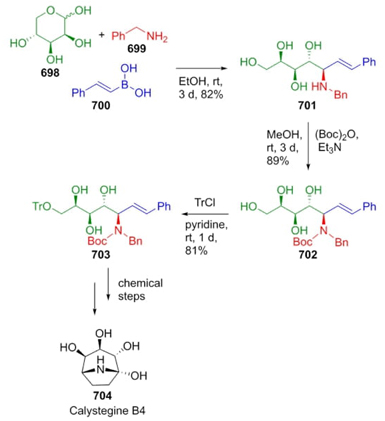
Hutton et al. reported a creative process to synthesize g,b-dihydroxyhomotyrosines through a Petasis reaction as one of keys steps [131]. Thus, glyoxylic acid (705) as carbonylic component, sulfinamide 706, and boronic acids 707 were combined to afford products 708 in good to excellent yields (90-99 %) and good diastereoselectivity. Then, compounds 708 were treated in concentrated acid media to give ammonium salts 709. Then, authors protected amino position to further transformations and completed preparation of dihydroxyhomotyrosine derivatives 710 (Scheme 107). As seen, the use of tert-butylsulfinamide in the Petasis reaction facilitated the incorporation of protective groups on the amine moiety. The olefin fragment was compatible with all conditions throughout the process, so it could be used for further transformations.
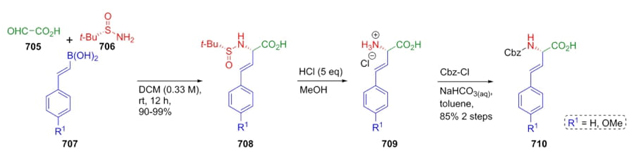
Thomson et al. reported an interesting methodology to provide allenes via a traceless Petasis reaction [132]. The reaction mechanism suggests that the first step is the formation of hydrazones 713 through the combination of sulfonylhydrazides 712 and glycoaldehyde (711). Then, the addition of alkynyl trifluoroborate salts 714 to hydrazones 713 gave propargyl hydrazides 715, which releases sulfinic acid to give diazines 716. Finally, loss of nitrogen molecule provided allenes 717 in moderate to good yields (61-90 %) (Scheme 108). Best yields were afforded under Lewis acid catalyst La(OTf)3 and acetonitrile as solvent. Alkyne was used as an activator of borate because the reaction does not work well with aldehydes that do not contain an α-hydroxyl group. This process may be considered as a reaction that did not follow the expected Petasis mechanism.
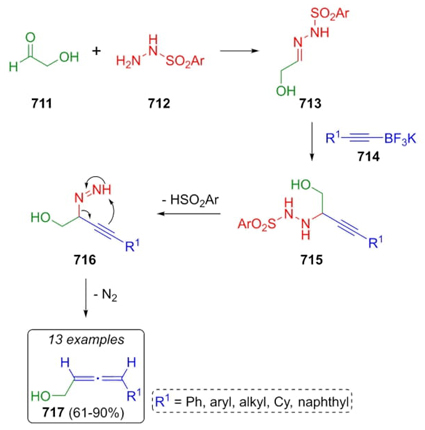
Shaw, Ghosal et al. reported in 2012 a creative methodology involving a diastereoselective Petasis reaction [133] from the carbohydrate galactose (718). Thus, iodide 719 was treated with Zn and Vitamin B12 in catalytic amounts to generate hemiacetal 720, which was combined in a one-pot process with trans-2-phenylvinyl boronic acid (721) and tert-butylamine to furnish the chiral alcohol 722, which after several chemical steps afforded the natural product (+)-Conduramine E (723) (Scheme 109). Petasis reaction was the key step to achieve in a diastereoselective way the key alcohol 722, as well as for reducing the number of synthesis steps.
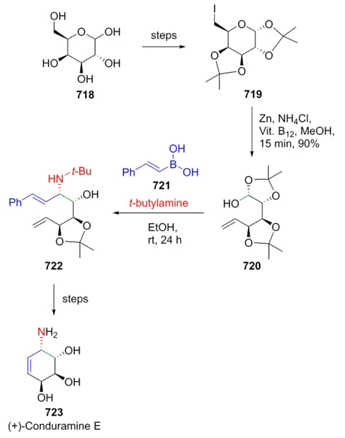
Yudin et al. reported a different approach to obtain aziridine-containing diamines via a Petasis-type reaction [134]. The mechanism proposed involves ring-openings in the substrates 724 toward aldehydes 725 as the first step. The addition of secondary amines 726 to aldehydes 725 in tautomeric equilibria with intermediates 727 promotes amine adducts 728, which in the presence of boronic acids 729 gave the hemiaminals 730. Thus, aqueous media afforded chelates 731. Si face attack is preferred over Re one to give the intermediates 732, which are hydrolyzed to yield products 733 in up to 72 %. Finally, a dimerization of the released compounds 734 may occur to recover the initial reagents 724 after losing an equivalent of boronic acid(Scheme 110). Products were synthesized in excellent diastereoselectivity (up to 95:5 ee). Through Petasis reaction, the key chelated intermediate 732 was assembled, which contributes in a high stereoselective way to form the syn products. On the other hand, the efficiency of the reaction depends on the halogenated solvent and the nucleophile used.
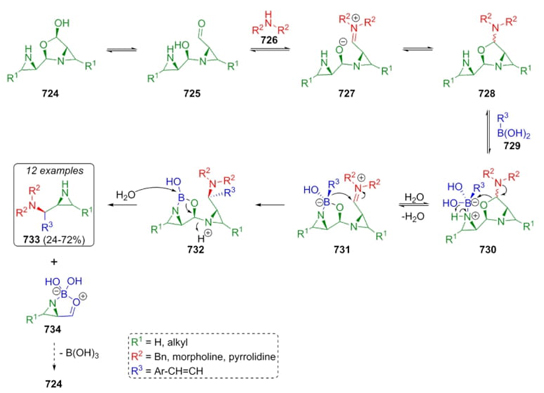
Around 2013, Shi et al. reported a version of the Petasis reaction in which salicylaldehydes 735, secondary amines 736, and vinylboronates 739 are combined sequentially under organocatalytic conditions to obtain the corresponding Petasis products in moderate to good yields (34-94 %) and in excellent enantioselectivity [135]. A plausible reaction mechanism suggests an initial formation of iminium ions 738 by condensation of salicylaldehydes 735 and amines 736. Besides, boronates 739 react with diols 740 and amines 736 under catalysis conditions to produce intermediates 741 in equilibria with 742, which were treated with preformed iminium ions 738 to afford the intermediates 743, which migrates a vinyl group after hydrolysis to finally obtain products 744 (Scheme 111). Another pathway in the formation of intermediates 746 was carried out through precursors 745 and iminium ions 738, which could be promoted by triethylamine in reaction media. Also, vinylboronates promote higher yields instead of other types of boronates such as dioxaborolanes or dioxaborinanes.
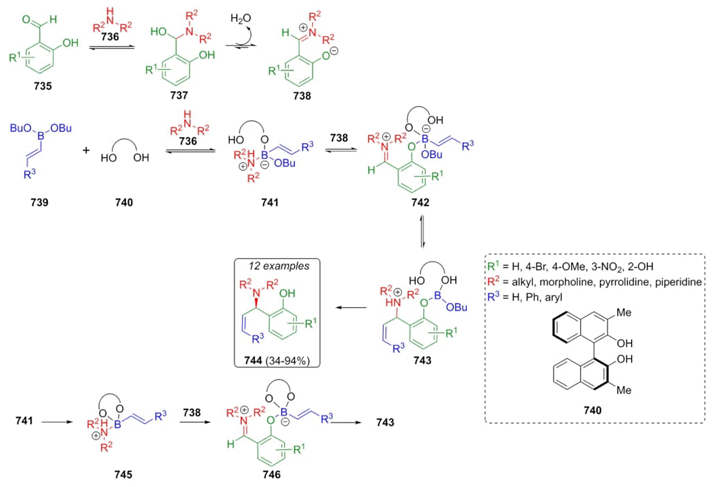
Seeberger et al. developed the total synthesis of legionaminic acid 754 using a stepwise methodology in which a Petasis reaction was the key step [136]. Synthetic procedure starts from D-threonine (747) (Scheme 112). After corresponding chemical transformations, diester 748 was treated with lithium borohydride to give the diol 749. Oxidation of compound 749 provided the less hindered aldehyde 750 as a reagent for Petasis reaction. Thus, a combination of aldehyde 750, amines 752, and styrylboronic acid (751) in ethanol afforded products 753 in 76 % yield as a diastereomeric mixture. All procedure was accomplished in eleven steps. Petasis reaction allowed obtaining products with good stereoselectivity, avoiding epimerization processes.
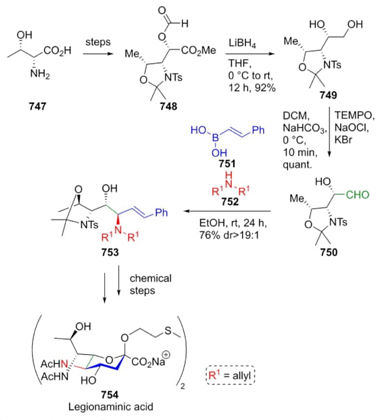
Clausen, Nielsen et al. reported a Petasis-based strategy to develop new scaffolds containing four stereogenic centers [137]. Thus, salicylic aldehydes 755, benzyl allyl amine (756), and furan-2-boronic acid (757) were reacted together to give the products 758 after a domino process Petasis / Diels-Alder cycloaddition. Then, cyclic compounds 758 were oxidized using K2OsO4 and methylmorpholine oxide affording the intermediate s759. Finally, products 760 (library B) were synthesized through a reductive cyclization promoted by osmium salt, NMO, and sodium periodate. Moreover, Library A was afforded after reduction of intermediates 761 coupled to cascade Mitsunobu conditions from precursors 762 (Scheme 113).
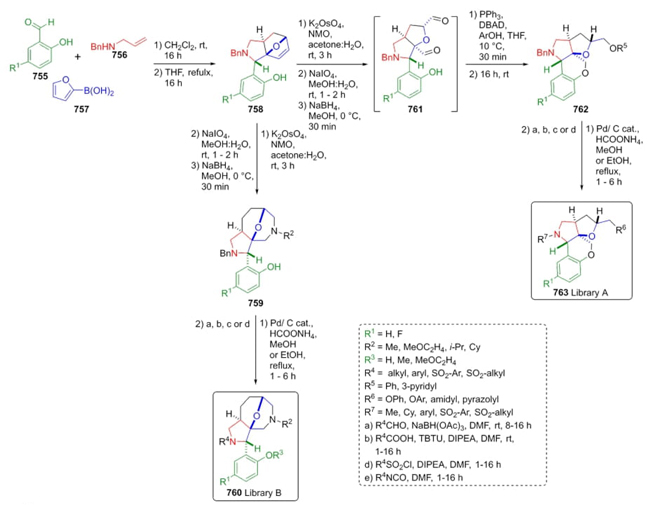
Wesjohann, Rivera et al. developed for the first time a synthetic procedure for labeling and stapling peptides via a new Petasis-type reaction without charge modifications [138]. The peptide chain was modified by protecting amino groups and linking an amino-end chain to lysine. Thus, the peptidic chain in polypeptide substrates 765 was reacted with aldehydes or ketones 764 and boronic acids 766 to afford peptide labeled on lysine side chain 767 (Scheme 114(a)). In the same way, a diversification of resin peptide was performed changing sugars such as D-erythrose or D-ribose, fluorescent peptides, or steroidal peptides instead of aldehydes. A Petasis-stapled reaction was carried out mixing resin-peptide 768, carbonylic compounds 764 or 769, and diboronic acids 770 to give peptides 771 (Scheme 114(b)). Due to polypeptide size, the authors carried out the MCR in two stages, to avoid a second Petasis reaction.
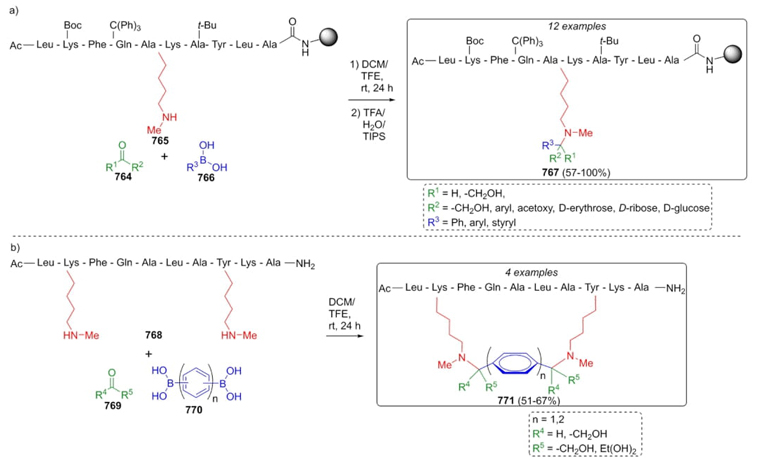
Zhang, Yang et al. reported a distinct procedure to obtain functionalized dihydropyrones via a regioselective Petasis reaction [139]. It proposed a mechanism pathway where iminium ions 775 were formed through condensation of glyoxylic acid (772) as a carbonyl compound and secondary amines 773 (Scheme 115). Oxaborols 776 were coordinated with carboxylates of 775 to achieve the intermediates 777. A further intramolecular nucleophilic attack of the vinyl moiety to iminium furnished intermediates 778, which after releasing a molecule of water achieved products 779 (78-98 %). Finally, thermal isomerization gave the products 780 in moderate to good yields (45-98 %). The authors found that its methodology cannot be applied using anilines as reagents as well as alkylic chains as substituents on the R3 position. Final isomerization processes are promoted by increasing temperature up to eighty Celsius degrees.
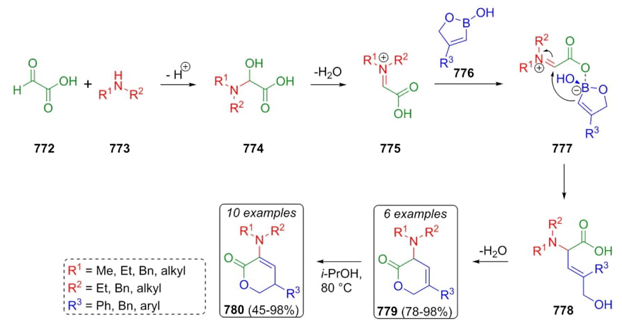
Bolm et al. reported in 2021 another approach to synthesize benzodiazepine analogues using a Petasis reaction under catalyst-free conditions as the key step [140]. With optimal reaction parameters in hand, glyoxylic acid (781), sulfoximines 782, and aryl boronic acids 783 were mixed without catalysis to afford products 784 in moderate to excellent yields (27-99 %) and diastereomeric ratio up to 1:1. Thus, product 784a was selected to perform further transformations involving esterification of its acid moiety assisted by trimethylsilyl diazomethane, and a further nitro reduction with iron in dust and acetic acid to give the precursor 785. Finally, its amidation with lithium hexamethyldisilazane promoted a ring closure to give corresponding benzothiadiazepine 786 in 55 % overall yield (Scheme 116). The authors noted that steric hindrance has no effects on the yields, except for S-alkyl substituent from sulfoximines. On the other way, electronic effects decrease the yields in alkyl-substituted S-aryl of sulfoximines.
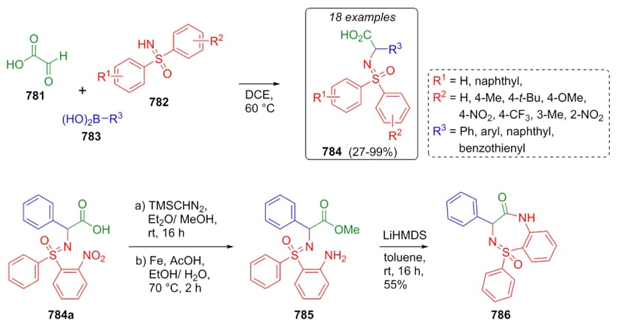
Povarov-type 3CRs
In 1967, L. S. Povarov reported the first reaction which involved a sequential combination of aromatic imines and alkenes reacting through an inverse electron demand Diels-Alder [4+2]-cycloaddition [141]. Later, this reaction was retaken and discussed into a 3-CR between aldehydes, amines, and olefins. The mechanism pathway is still under discussion, as the first step is a condensation between aldehydes 787 and amines 788 to give corresponding imines 789 in catalytic conditions. Then, two routes have been proposed (Scheme 117): a) via a Mannich addition to alkenes 790 to achieve the intermediates 791, followed by Friedel-Crafts alkylation, and finally an aromatization step to produce the compounds 793; and b) same route Mannich addition and Friedel-Crafts alkylation were carried out to construct the concerted intermediates 794, which react through a cycloaddition-type reaction to give the products 793 [142].
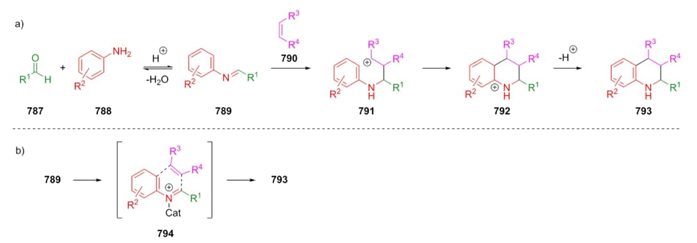
In 2015, Muñoz-Torrero et al. reported the synthesis of quinoline-derivatives using a Povarov reaction as a first key step synthesis and their biological evaluation against protozoan parasites T. brucei, T. cruzi, and L. infantum [143]. The synthetic strategy of the more active compounds begins with a Povarov reaction between corresponding aldehydes 795 and amines 796; in the case of the olefinic compounds 797, lactams, enamines, or pyrans were used. Further transformations such as oxidation with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) reagent, protection/deprotection processes, and reduction of nitriles or lactams were carried out in each sequence (Scheme 118). To improve the activity of the compounds as well as higher bioavailability, the authors proposed a bioisosteric replacement of NH to O or the incorporation of heterocyclic rings instead of cyclic amide.
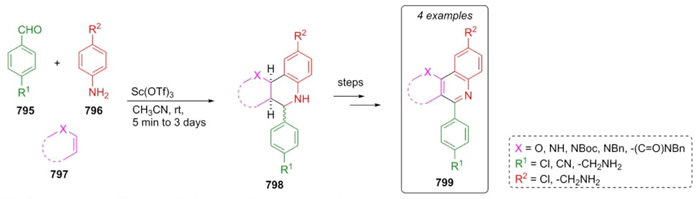
Menichetti et al. reported in 2019 the synthesis of heterohelicenes via the Povarov-oxidation process through a one-pot manner [144]. A combination of benzaldehyde (800), naphthylamines 804 or anilines 801 and 805 , and as cyclic aromatic dienophiles, alkenes or chromenes 806, 807, and 808 respectively afforded the products 803 or 809, under different reaction conditions. Reaction conditions a) it was carried out using boron trifluoride ethyl etherate as a catalyst to achieve a diastereomeric mixture cis/ trans of tetrahydroquinoline 803 in moderate yield. Then, under reaction conditions b) scandium triflate or ytterbium triflate were used as catalysts in different amounts, magnesium sulfate as an additive, and DDQ as an oxidative reagent to achieve the compounds 809 in moderate to good yields (34-75 %) (Scheme 119). The authors proposed that involved imines act as dienes to afford the compound 803 (depending on the amount of amine) but also participates into oxidations to give compounds 809. Naphthylamine directs towards products 809 regioselectively due to a-carbon or b-carbon that participates in cyclizations.
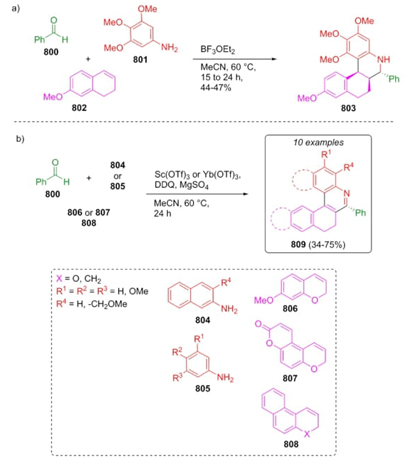
Kuwabara et al. reported in 2021 the synthesis of some new triazatriphenylenes via a double Povarov reaction [145]. Aromatic aldehydes 810, dianiline 811, and phenylacetylene (812) were sequentially combined under Boron trifluoride etherate as a catalyst, and DQQ as an oxidative reagent to afford a mixture of products 818 and 819, being 4,7-phenanthroline derivative 818 the majoritarian product (52 %). Authors proposed a mechanism pathway where the first step is the Povarov cyclization under a Lewis catalyst to give the compound 813. Then, the addition of acetylene 812 achieved zwitterionic compounds 814, which through [1,3]-H shift afforded the compounds 815. Compounds 818 were obtained after a ring closure, prototropic tautomerization, and oxidation (Scheme 120). To know about the second cyclization, DFT studies were performed and showed that in compound 815 there is a HOMO charge distribution in the a-position of the quinoline core that favored the cyclization. Also, steric repulsion from the phenyl neighboring fragment did not interfere with the cyclization process.
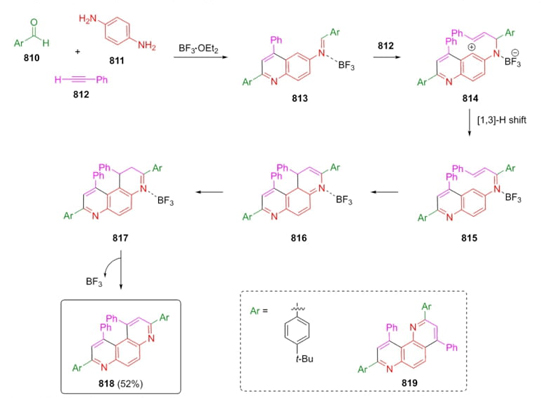
Wu et al. reported the synthesis of various new tetrahydroquinolines in 2022 which were evaluated behind their interactions with micro RNA-binding protein LIN28 and let-7 micro RNAs, both related to human tumor cells. After finding optimal conditions, a combination between aromatic and non-aromatic aldehydes 820, anilines 821, and cyclopentadiene (822) using scandium triflate or ytterbium triflate as catalysts achieved tetrahydroquinolines 823 in moderate to good yields (21-94 %) and diastereomeric ratio (up to 99:1). Based on the stereoelectronic properties of the anilines 821, mixtures of cis/ trans products or syn products as major diastereomers were obtained. Authors were inspired for this work in the compound LI71, which was previously reported [146]. The goal of synthesizing these molecules is to have greater interactions with the non-coding RNA molecules LIN28 and let-7.
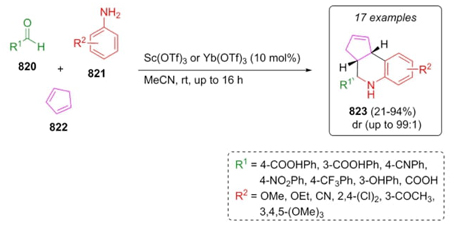
Thomas et al. reported in 2024 the synthesis of a covalent organic framework (COF) via a Povarov reaction [147]. Sequential combination of aldehyde 824, aniline 825, and styrene (826) afforded the COF 827 under Lewis acid catalysis and DQQ as oxidant in good yield (75 %) (Scheme 122). COF 827 exhibited stability under strong acids and bases. Povarov reaction allowed affording a complex product in one step. Also, the porous and crystalline rearrangement was favored due to Povarov reaction being performed after the oxidation process resulted in a π-π stacking interactions of extended frameworks.
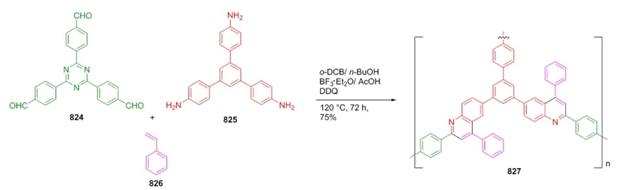
Miscellaneous
Unnamed 3CRs
Katsumura et al. reported the first asymmetric total synthesis of (-)-Hippodamine by an unclassified three-component reaction as the first step [148]. Thus, after optimal conditions were found, a sequential combination of aminoalcohol 828, ester 829, and vinylstannane 831 under palladium catalyst achieved the product 834 in 81 % yield as single diastereoisomer (Scheme 123). It is proposed a mechanism pathway where reagent 828 in the presence of ester 829 allowed the formation of compound 830. Then, an addition of stannane 831 occurred to promote further aza-electrocyclization to give compound 833. Further sequential transformations such as hydrogenations, alkylations, and intramolecular Mannich reactions completed the synthetic methodology toward the natural product 837.
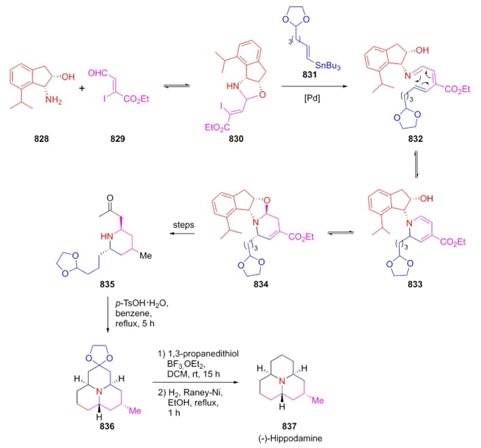
In 2019, Huang, Deng et al. developed a synthetic route to obtain naphthothiazoles 846 via a three-component reaction under catalyst and additive-free conditions [149]. A plausible reaction mechanism was proposed, where tautomerization of cyclohexenone oximes 838 provided the enamines 839 (Scheme 124). Then, elemental sulfur was attacked probably via a Willgerodt-Kindler process by compounds 839 to give the intermediates 840, which were annulated after the addition of aldehydes 841 to give intermediates 844. After releasing water and acetic acid, the intermediates 845 were obtained. Finally, it was oxidized with sulfur present in the medium affording the products 846 in moderate to good yields (31-93 %).
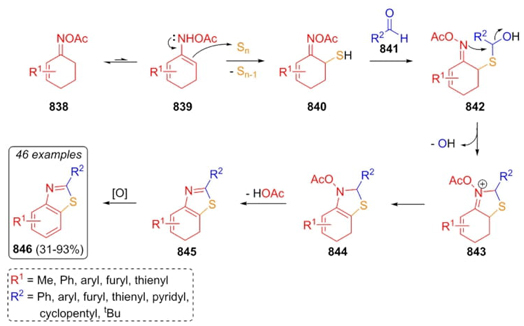
In 2019, Lomas-Romero, Gonzalez-Zamora, Islas-Jacome et al. developed a procedure to synthesize a few symmetrical tetrazoles 849 through a non-named 3CR involving a heterocyclization as a key step [150]. Thus, a sequential combination of anilines 847, trimethyl orthoformate (848), and sodium azide gave the products 849 in moderate to good yields (30-83 %). The plausible reaction mechanism starts with the formation of activated carbonyl intermediate 851 from trimethyl orthoformate (848) (Scheme 125). This latter reacts with the amines 847 via a condensation reaction to give the dipoles 854 after releasing a molecule of methanol. Then, these later intermediates carry out a dipolar cycloaddition with azide anion to assemble the symmetrical tetrazoles 849. These compounds are potential linkers to fabricate tetrazole-based MOF-type materials. Tetrazole as a functional group is considered an isostere of carboxylic acid. In this context, tetrazole-based ligands have some advantages over carboxylate-based MOFs, like enhanced resistance to harsh conditions like higher temperatures, a wide range of pH, and a higher degree of humidity.
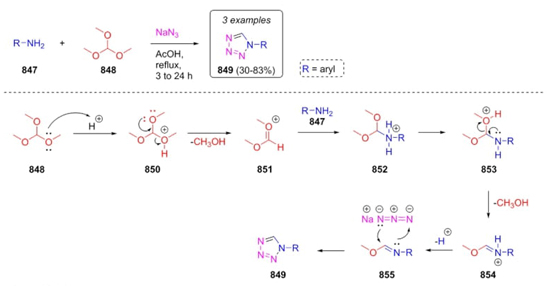
Computational calculations on 3CRs
Over time, it is commonly found collaborations between experimental and theoretical chemists behind searching mechanism pathways. This one provides greater robustness to the work reported. In this subsection, we discuss some works describing energy profiles through computational studies.
One of the most controversial MCRs without using isocyanide is the Biginelli reaction, whose mechanism is still under discussion. In 2015, Morokuma et al. reported mechanistic studies about the most favorable pathway in the Biginelli reaction between benzaldehyde, urea, and ethyl acetoacetate [151]. It is necessary to mention that urea performed a double function as a reactant and catalyst. The three routes of Biginelli mechanism (iminium, enamine, and Knoevenagel pathways) were evaluated under artificial force-induced reaction method computed using DFT formalisms at M06-2X/6-31+G(d) level of theory, in ethanol as the best solvent, Fig. 1.
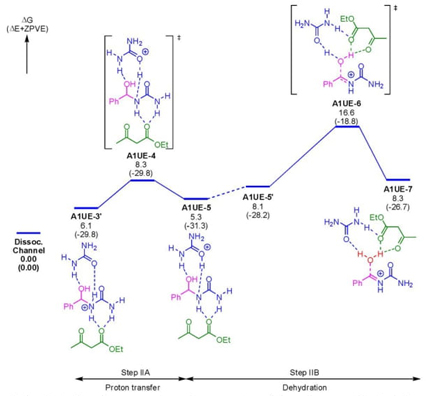
Fig. 1 Revised mechanism of the Biginelli reaction determined by the artificial force-induced reaction method. Reproduced from ref 150. Copyright 2015 American Chemical Society.
In Mannich-type reactions (previously discussed) [60], a combination of dienediolates and imines follows a pathway giving stable intermediates produced after a Pictet−Spengler-type cyclization. A nice energy profile was computed by Schneider et al. through DFT formalism using M06-D3/LACVP**/PBF method including London dispersion interactions using acetonitrile as the best solvent, Fig. 2.
Conclusions and outlook
Every year several examples of multicomponent reactions without using isocyanides are published, mostly emphasized in "most popular" three-component reactions like Strecker, Biginelli, or Mannich. However, in the last ten years, many other examples “less common” like Petasis, Reissert, Ritter, Gewald, Povarov, Bucherer-Bergs, Willgerodt-Kindler or Kabachnik-Fields have gained their importance in various fields of science and technology. Thus, the present comprehensive review is focused on medicinal applications, total synthesis, green conditions, and novel mechanisms (or variants) proposed, directed towards experts and those who are not specialized in multicomponent reactions, but from different areas of chemistry.

