











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction and generalities
The intend of this review is to survey the contributions developed in the research group of prof. Dr. Hiram Isaac Beltrán during the last 22 years of his career, 3.5 years at Instituto Mexicano del Petróleo, 1 year at Instituto de Química UNAM, and the last 17.5 years at Universidad Autónoma Metropolitana, first in Unidad Cuajimalpa and later in Unidad Azcapotzalco. During this period, research was carried out to develop i) organic ligands to obtain ii) successive boron and tin coordination compounds with them, also investigated iii) macrocyclic chemistry systems of phthalocyanines and porphyrins, iv) polymer synthesis and functionalization, v) supramolecular synthesis, vi) coordination polymers and metal-organic frameworks (MOFs), vii) among other systems. All this research involved the design and development of a particular molecular or material system with specific physicochemical properties to achieve a goal or a desired property or set of properties, to eventually fulfill a function or be functional or even multifunctional for biologic, scientific or industrial purposes. To follow all these lines of research, some concepts will be highlighted in the next sections and later the compendium will be presented. The acquaint of this knowledge and experience has made it possible to design and develop molecular and material systems to achieve specific functions and applications. This has also helped other research groups to understand their own studied systems and has furthered the research field of applied chemistry at a multifunctional level.
Structure ↔ Property/Activity Relationships
In the chemical sciences, one of the most important principles is that the structure of molecules, compounds or substances is directly responsible for their physicochemical properties and biological activities. In this sense, one of the author's workgroup main goals was to understand the structure of these chemical systems in order to relate them to their properties, functions and activities, and finally to understand the context behind this structure↔property relationship or linkage.[1-6] This ultimately helps in the design, engineering, and development of functional molecules or materials for potential applications to incorporate the required functional groups into a predefined chemical architecture to achieve the desired physicochemical properties. When a scientist or engineer is able to systematically go through this process, (s)he reaches the level of applied chemistry by moving from the level of complexity to the level of coherent understanding, where the term “complex” stands for something incomprehensible and the term “coherent” stands for something logically, cleverly or intelligently ordered or integrated with the precise features to accomplish a specific task. E.g. Mother Nature develops its natural processes at the coherent level of understanding or conceptualization in a kind of green chemistry.
Structural features derived from characterization techniques
But here the question arises of how to recognize the atomic or molecular properties that these chemical substances have or need to perform the proposed tasks so that they are functional or could be applied? The typical way to collect structural information at this level, is therefore the use of characterization techniques. They are (multi)nuclear magnetic resonance spectroscopy (NMR), Fourier transform infrared spectroscopy (FTIR), UV-vis spectroscopy, single crystal and powder X-ray diffraction (SCXRD/PXRD), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), isothermal titration calorimetry (ITC), scanning electron microscopy (SEM) and others. They have all been the “eyes” and “hands” to see or track the phenomena at the micro, nano and even atomic and molecular levels of matter. [7-12] So, let’s examine the prerequisites for understanding the chemical and material systems at these levels. This means that we need to obtain chemical information about the constitution, the conformation and finally the configuration, meaning that these three levels of chemical information are linked to the general chemical knowledge of matter.
Coordination compounds and the need to synthesize appropriate ligands for coordination purposes
An important combination of information could be acquired as an organic chemist, or as an inorganic chemist, but as a coordination chemist, this means a chemist having the two viewpoints at once. This inorganic-organic synergy has proven useful in drawing attention to vital phenomena and understanding the behavior of these molecular/materials systems. Coordination compounds could be considered as a link between organic and inorganic chemistry, as they deal with the interaction between metals and organic molecules. It is precisely this type of information that has contributed to the development of other areas of chemistry such as supramolecular, bioinorganic, biophysical, etc. For the point of view of the coordination chemist, we should remember that the organic molecule serves as Lewis base and the (semi-)metals serve as Lewis acids, and that the vast majority of these reactions are carried out due to the template effect caused by the coordination of the organic molecules with the metallic center. This process crowds the boundaries of the metal with molecules and is considered a highly convergent chemical transformation that has led to a huge number of reactions and highly efficient synthetic conditions.[13,14] In this sense, the organic chemist plays a crucial role as they must have the right organic molecules to serve as a ligand, but organic chemists have their own chemical tasks and systems to develop. Thus, a coordination chemist not only has concepts of inorganic chemistry, but also the ability of organic synthesis to provide important chemical substances that are able to interact with metals to achieve the desired properties in the final coordination compound.
Coordination chemistry concepts applied to other scientific fields
Therefore, the scientist able to gather the coordination chemistry concepts, including organic and inorganic chemistry baggage, could sightsee across other scientific fields to understand them in a more accurate way. e.g. protein folding, metalloenzymes, supramolecular chemistry, interaction of light with matter, optical properties, luminescence, ground and excited states, materials chemistry, materials sciences, molecular physics, and nanosciences, including the so well-known terms bottom-up and top-down approaches.[13-24] Therein the coordination chemist would “see” the interactions between metals and specific functional groups in proteins and oligopeptides, understand the mechanistic level of catalytic processes like those happening in hetero/homogeneous systems. But also, by the use of (metallo)enzymes, as well as the doping of metallic elements or ceramics, the synthetic approaches to obtain particular molecules or materials. e.g. the sol-gel method to obtain aluminosilicate porous materials and related compounds, perovskites, host-guest chemistry, coordination polymers, including metal-organic frameworks (MOFs), etc. As we have previously stated, all these systems could be prepared with the understanding and applicability of two major concepts: A) Template effect; and B) Self-assembly. Both terms are commonly applied in the fields of coordination chemistry, supramoelcular chemistry, and materials chemistry, because of the effects that they cause over the final structures.
Template effect concept
The template effect could be principally cornered when the metallic centers gather Lewis bases present in organic moieties with free electron pairs. By the formation of coordinative bonds, the metallic center is able to assemble or template a particular coordination compound with the organic ligands in a highly convergent, multicomponent, synthetic approach.[25-27] This happens due to the closer interaction between the Lewis acid, the metallic atoms, as the structural center, with the Lewis bases, as the surrounding neighbors, forming a new molecular entity that is the coordination compound. This is not the only case where a template effect is present but is the most known. This also occurs when anionic or cationic molecular species are present, and indeed due to their own structural and physicochemical properties, could pre-organize a particular molecular entity.
Self-assembly concept
The self-assembly is the preorganization of molecular or atomic entities, that could remain in that supramolecular state, where weak interactions play the initial role to generate such structure. But if they undergo a chemical reaction, they are now a part of a larger molecular entity, nevertheless they passed through the self-assembly state, to preorganize the final structural entity. Precisely by this preorganization the final system could be obtained in a very convergent approach. [25,27-29]
Since one of the first contributions, in 2003, [30] we could be able to prepare diorganotin(IV) coordination compounds by template effect and derived dimers, trimers and even coordination polymers by self-assembly, this was achieved by intermolecular coordinative bonds formation.[31,32]
The template effect is strongly present, not only in normal coordination compounds, and other supramolecular assemblies, but also in the formation of more complex systems. Indeed this is one of the key concepts to develop the coordination polymers and MOFs systems, [26] the other is the self-assembly, both of them are direct rensponsible for the formation of these very interesting materials. [25]
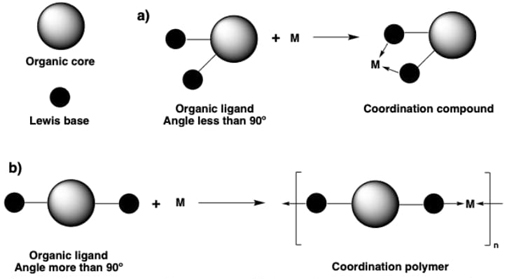
Coordination polymers (CP) and metal-organic frameworks (MOFs)
A coordination polymer (CP) is a superlative of a coordination compound, the main difference is that the organic ligands were constructed or selected in such a way to build coordination moieties (adducts, chelates, or even macrocycles) across various “directions”. This has prompted to the obtaining of coordination polymers of various dimensionalities starting in 1D, passing by 2D, and ending at 3D.[33-35] The main key herein is to have at least two Lewis bases sites available in the same chemical entity, and some of these Lewis bases should be strategically placed along the organic backbone in obtuse (at least 90⁰) geometrical angles, preferably approaching at 120 and 180⁰. This principle allowed to link one coordination moiety with another, and giving place to the coordination polymer in one, two or three spatial directions, see Fig. 1. In particular, the metal-organic frameworks (MOFs) are a subclass of coordination polymers, and the principal characteristic of MOFs is that this type of compounds present permanent porosity, thus they were also named porous coordinate networks (PCN). In this type of research areas converge many other areas of chemistry and materials sciences, being the coordination chemistry the closest or central area/discipline to understand the occurring phenomena.
Results
Developed research in the chemical and materials sciences
With this background acquired during the research experience over the years, the next sections will now review the synthetic methodologies designed/engineered to produce:
Organic ligands used to obtain boron-coordination compounds,
Diorganotin(IV) coordination compounds directly obtained by template effect,
Applications and research lines derived from the developed coordination compounds of boron and tin,
Coordination polymers and metal-organic-frameworks (MOFs),
Infiltrated coordination polymers and MOFs,
Bandgap modulation and luminescent properties of coordination polymers and MOFs,
Biological properties of coordination polymers and MOFs,
Macrocyclic systems and their applications, and
Supramolecular assemblies and their applications.
All these compounds, no matter of their molecular or materials nature, were accompanied by structural and physicochemical characterizations such as NMR, FTIR, UV-vis, TGA, DSC, ITC, SEM, and single crystal and powder XRD, among others.
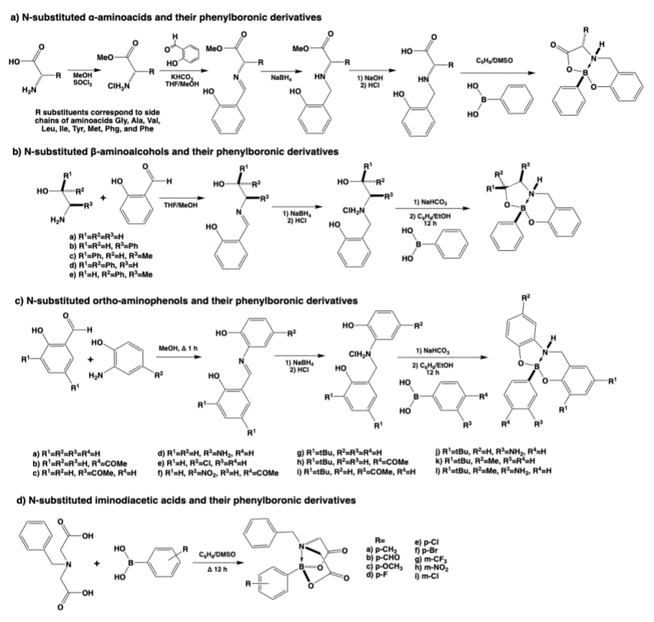
Organic ligands used to obtain boron-coordination compounds
Some of the first attempts to obtain organic ligands to develop coordination compounds resulted in publications of substituted α-aminoacids, β-aminoalcohols, or ortho-aminophenols some of them first presented in this same journal 22 years ago,[36] Fig. 2. The obtained ligands were generated by analog synthetic methodologies [36-38] starting from the α-aminoacids, β-aminoalcohols, or ortho-aminophenols which are already mono-chelating (bidentate) ligands and provide another coordination arm to obtain bis-chelate ligands or tridentate, therefore with an augmented chelate effect to improve the coordination ability and stability. The coordination compounds obtained with these ligands were firstly boron derived species, Fig. 2(a-c). [37, 39] Also some years later the workgroup developed another family of ligands, named amino-bis-phenols, obtaining again boron coordination compounds with these new tridentate ligands, named boronate-amino-bis-phenols (Fig. 2(c)). [38]
Another interesting set of coordination boron compounds could be obtained in the workgroup giving place to boron iminodiacetic esters, named (N→B) phenyl substituted[N-benzyliminodiacetate-O,O’,N]boranes. All these compounds exhibited fused bicyclic structures because of the formation of an intramolecular N→B bond, and the stereochemistry at the fusion, again resulted in a syn disposition. A simple but interesting structure-property correlation between δ(11B) and σHammett values according to the substituents attached to the aromatic ring of the phenylboronic moiety revealed that the strength of the N→B bond depends on the electronic factors of the substituent at this B-Ph group, see Fig. 2(d). [40]
Diorganotin(IV) coordination compounds directly obtained by template effect
By understanding the template effect, which is often employed in the synthesis of coordination compounds, some other systems could be investigated. Therefore, we would like to point out that the common approaches used in the literature to obtain diorganotin(IV) coordination compounds were two- or three-step synthetic methods that involved first synthesizing the organic ligand and later reacting this ligand with the diorganotin(IV) moiety. [41]These common approaches yielded coordination compounds with 5, 6 or 7 coordination numbers at the tin center and used the rather insoluble diorganotin(IV) oxides (R2Sn(IV)O), or the diorganotin(IV) dihalides such as R2Sn(IV)Cl2, where usually R=Me, nBu or Ph. [41]Some other approaches dealt with the use of more basic salts such as Sn(acac)2 or SnCl4. All tin(IV) starting materials containing halides required the preactivation of the organic ligand with a base such as NaOH, NaHCO3, etc., which was also used to neutralize the formation of e.g. HCl during the course of the reaction in situ and to obtain NaCl by-product, that should be separated in such a way. In some cases, N-containing bases (e.g. triethylamine, NEt3,[42] or pyridine, py) could also be used, as is common in organic chemistry, but with caution due to the possible formation of Sn(N adducts that could interfere with the formation of the desired coordination compound. All this is done for the simplicity of the synthetic approach, without considering the additional electronic properties that could accelerate/ralentize the reaction kinetics and confer particular physicochemical properties to the final compound. Nevertheless, one of our first contributions is the one in which we were able to realize a direct one-pot synthetic approach without using all these previous strategies, starting from the raw components, in this case the diorganotin(IV) oxides (R2Sn(IV)O), and those that should yield to the organic ligand in situ. [30]A very important point is that all these starting materials could be transformed by condensation reactions, generating in situ the iminic tridentate bis-chelate ligands to obtain the desired diorganotin(IV) coordination compound. In this first approach, not only the quite insoluble diorganotin(IV) oxides (R2Sn(IV)O) were used, but also the also quite insoluble L-amino acids. [30]However, once a small amount of the coordination compound was formed, it was so stable that the reaction was directed towards the products, shifting the equilibrium in moderate to high yields. Using this simple one-pot strategy, several diorganotin(IV) coordination compounds could be prepared, including 3, [30-32,43-45]4, 5 [46]or even 8 [47]components, in a highly convergent synthetic method that always gave a reaction yield of >50%. And all this was achieved without any further purification steps, as the water formed during the multiple condensations could be displaced from the reaction medium by using a Dean-Stark trap.
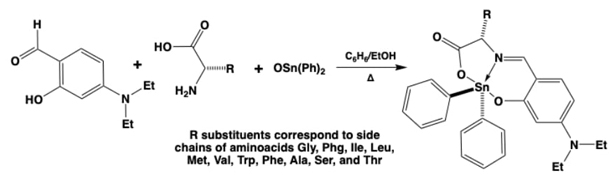
So, with these chemical advantages provided by metals and semimetals, three or more component convergent synthesis were designed to obtain dioroganotin(IV) coordination compounds derived from aminoacids,[30] β-aminoalcohols, [32]and ortho-aminophenols (Fig. 3),[31] in situ providing the iminic-tridentate ligands by multi-condensation reactions in all cases, see Figures 3-7. Further studies were carried out with some of these compounds, since it is well known for the organotin derived molecules to have biological properties. They behave in an analogue way to anticancer drugs such as cis-platin and other metallotherapeutics. Nevertheless, the organotin(IV) based compounds have also revealeded biocide (mainly antibacterial) and anti-fungal capacities. [43]Therefore, the ortho-aminophenols derived family presented interesting bactericidal activities against Gram-positive/negative subtypes and anaerobic bacteria. [31]Also, these coordination compounds were tracked to assess their further environmental impact by the determination of their acute toxicity using LBT-Microtox tests (luminescent bacteria toxicity), determining that this activity could be modulated through the proper selection of the disubstituted tin moiety.
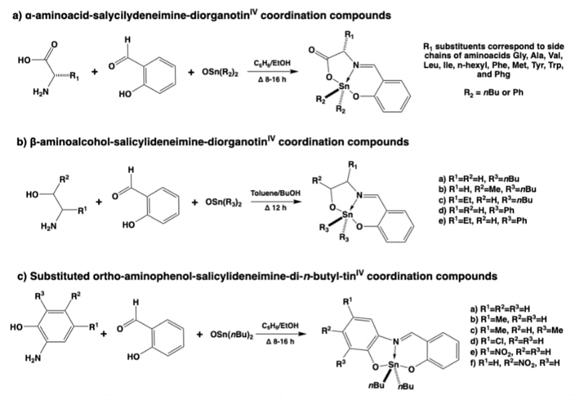
Fig. 3 Synthetic strategies to yield dibutyl and diphenyl-tin(IV) coordination compounds by the in-situ assembly of iminic tridentate ligands derived from (a) α-aminoacids, (b) β-aminoalcohols, or (c) ortho-aminophenols.
Due to the observed biological activities that these tin coordination compounds revealed, some other synthetic routes were surveyed to yield diphenyltin(IV) coordination compounds derived from substituted ortho-aminophenols (Fig. 4). [44]These compounds were tested in vitro against six human tumor cell lines assessing their antitumor activity; besides they were also tested for their antioxidant efficiency in rat brain homogenate. Structure activity correlations were performed to determine the principal structural features responsible for such activities, seeming to be an inverse structure-response behavior among activities, since the less active compound for cytotoxic assays resulted the best in antioxidant tests.
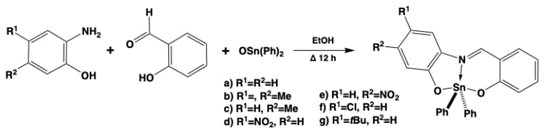
Fig. 4 Synthetic strategies to yield diphenyltin(IV) coordination compounds by the in-situ assembly of iminic tridentate ligands derived from substituted ortho-aminophenols with multifunctional biological activities.
Also due to the important structural characteristics of diorganotins, the research group investigated the photophysics of 14 diphenyltin(IV) coordination compounds self-assembled with the Schiff bases of salicylidene and 2-hydroxy-naphthylidene ortho-aminophenols obtained in situ (see Fig. 5). [45]These coordination compounds revealed a 50-fold increase in excited-state decay times compared to free iminic ligands. This high stability enhancement is directly related to the coordination of this ligand to the metallic center, freezing rotational fluctuations passing from the free-ligand to the coordinated-ligand. Another important result is that the emission bands of these coordination compounds could be fine-tuned by placing substituents on the organic backbone. This highly enhanced emissive behavior facilitated the investigation of the properties of electronically excited states by correlating the effects of different substituents with the resulting femtosecond and picosecond dynamics. In addition, the two-photon absorption cross sections (TPACS) were determined by measuring the two-photon induced fluorescence excitation spectra in the 760-820 nm spectral range. All the obtained results indicate that the diphenyltin(IV) structural fragment, due to its large metallic size and pentacoordinate mode assembled with the chromophoric Schiff bases, are conjoint structural prerequisites through which the emissive states developed lifetimes in nanosecond time scale.
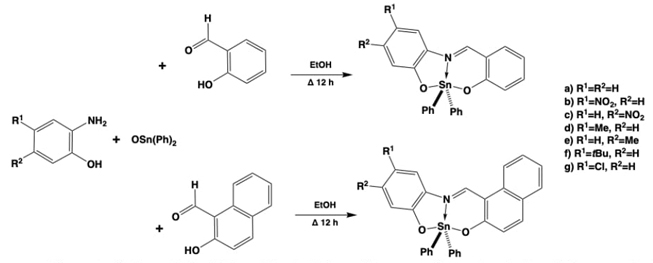
Fig. 5 Self-assembly of diphenyl-tin(IV) with iminic tridentate ligands derived from substituted ortho-aminophenols.
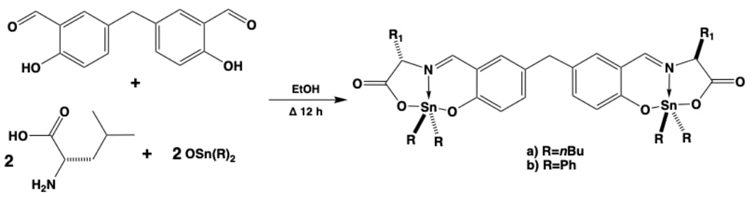
Lately, two bis-di-organotin(IV) compounds were prepared in good yields (ca. 80 %) by a multicomponent one-step five moieties self-assembly reaction through the interaction of 2 eq. of aminoacid leucine, 1 eq. of 5,5’-methylene-bis-salicylaldehyde, and 2 eq. of di-n-butyltin(IV) or 2 eq. of diphenyltin(IV) oxides, providing these bis-di-organotin derivatives, see Fig. 6.[46] The new coordination compounds were tested as corrosion inhibitors in sour brines on mild steel surfaces, presenting inhibition efficiencies of 54 and 68% for butyl and phenyl organotin moieties, respectively. Importantly, compound bearing the (Ph2Sn) moiety developed a slightly better inhibition, suggesting that this compound interacted more efficiently towards the metallic steel surface. This interaction could be assessed due to p-bonding interactions with the metal atoms, but also due to the formation of (O(δ-)-Sn(δ+))···(Fe(δ+)-O(δ-)) complimentary Lewis pairs, in a similar fashion as has been observed when dimeric/oligomeric diorganotin(IV) compounds self-assembled. [31,48] Even, this occurred strongly with the Ph2Sn moiety, due to a diminishment of steric strain, this in comparison with the nBu2Sn moiety, where the nBu fragments developed more strain because they have a bigger steric cone. This along with the contribution of the side chain fragment of leucine, conferring a hydrophobic barrier against the aggressive sour brine media, hence increasing and strenghtening the protected surface area. Since some other related compounds were tracked for their antibacterial capacities, these two species were tested in vitro against Bacillus subtilis (Gram-positive), Escherichia coli (Gram-negative), and Pseudomonas fluorescens strains, being more effective for the E. coli strain, at concentrations as low as 25 mg·L-1. These two bis-coordination compounds were also tested for their asphaltene inhibition/dispersion activity in n-heptane media, being intended to be used as possible chemical stabilizers of petroleum and derived industrial currents possessing high molecular weight aromatics such as resins and asphaltenes. The obtained results strongly suggest that the presence of the four n-butyl moieties and the general bis-coordinative backbone with a core possessing two aromatic rings, are providing as a conjoint a good amphiphilic balance to obtain good dispersion efficiencies in the tests.
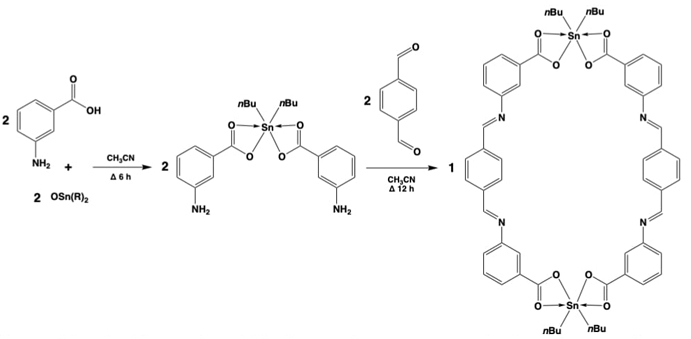
The workgroup could also obtain macrocycles by a self-assembly of organotin fragments with Schiff bases. [47]This has been identified as metallosupramolecular chemistry, carried by a hexacondensation process in two following reactions. The first step to construct the coordination moiety, by the 2:1 condensation reaction of 3-aminobenzoic acid with di-n-butyltin(IV) oxide in acetonitrile providing the diorganotin(IV) building block. The second step through the [2 + 2] condensation reaction with terephthalaldehyde and the already obtained diorganotin(IV) building block, yielding 4-imine bonds at once, in a one-pot, 8-component process. All carried on in a one-pot strategy, to prepare a 38-membered bis-diorganotin(IV) macrocycle, of rectangular shape, with ca. dimensions of 17.4 Å (length) × 14.0 Å (width), see Fig. 7.
Applications and research lines derived from the developed coordination compounds of boron and tin
The research has aided or guided other researchers, therefore has been cited by diverse brotherhood workgroups and have applied related systems for diverse subjects, as will be reviewed following.
Since the obtained boron coordination compounds (see Fig. 2) generate two new stereogenic centers, one in N and one in B, with fixed stereochemical control being the N(B bicyclic fusion always in a syn disposition, derived compounds have been employed as asymmetric synthons, and the “boron based enantiomerism” concept has been acquainted. [49]Indeed, four-coordinate organoboron derivatives have presented interesting chemical, physical, biological, electronical, and optical properties. Due to an increasing demand to yield smart functional materials, these chiral organoboron compounds could be employed for this means, for the stereoselective synthesis of themselves stereo-boron organo-derivatives, or as chiral auxiliars for other transformations.[50]Some other examples revealed that this boron coordination compounds could be isolated as the pure enantiomers. e.g., the racemic boronate-imine and boronate-amine coordination compounds, both featuring a tetrahedral and thus stereogenic boron atom, were obtained starting from 2-amino-1,1-diphenylethanol with the corresponding phenylboronic acids. The enantiomers were separated, and the racemization barriers at the boron center were found to be in the same range for both amine and imine coordinations at ca. 100-110 kJ·mol-1. [51] Other research group has developed an enantioselective Cu-catalyzed dissymmetric B-H bond insertion procedure for the yielding of a series of boron-stereogenic molecular systems. This has been achieved starting from the already N(B tetracoordinated 2-arylpyridine-boranes with diazo compounds, generating this series of enantio-enriched boron-stereogenic compounds in moderate to good yields, presenting high enantioselectivity and diastereoselectivity.[52] Experimental evidence and DFT calculations suggest that the inclusion of a substituent at the ortho (B-related) position on the aromatic ring of the starting boranes is primarily responsible for the stereogenic control of this transformation. A further experimentation taking advantage of the already found procedure, was obtained for the 2-arylpyridine-borane which twice underwent B-H bond insertion propitiating a species with 3-carbon substituted boron-stereocenters.
Also related to this subject (Fig. 2(d)), [40]other research groups developed a simple and modular strategy for small molecule synthesis, through the employment of this boron iminodiacetic esters as synthetic scaffolds, by an iterative Suzuki-Miyaura (SM) coupling of B-protected haloboronic acid building blocks, that are able to dramatically simplify the otherwise quite complex coupling synthetic process. [53]These colleagues employed the commercially available bis-chelating ligand, N-methyliminodiacetic acid (MIDA), so a variety of haloboronic acids were N(B coordinated with the MIDA ligand to yield a series of B-protected bifunctional building blocks by this means.
For the case of the self-assembled diorganotin(IV) coordination compounds, this simple strategy allowed other workgroups to obtain their own organotin(IV) derivatives. e.g., the assembly of tetradentate ligands by the 1:2 reaction of ortho-phenylenediamine with substituted salicylaldehydes gave N,N-bis(2-hydroxy-4-R-benzylidene)-1,2-phenylendiimine. These ligands were coordinated with diorganotin(IV) moieties (Me, n-Bu, Ph) and conducted to the formation of six new organotin(IV) coordination compounds with distorted octahedral hexacoordinated tin(IV) atoms, see Fig. 8. Due to the high π-conjugation but also to the structural fixation of the systems while coordinated with tin center, these compounds were tested for their non-linear optic (NLO) capacities, exhibiting reasonably good quadratic NLO properties. This has been seen because of the obtaining of interesting μ·β (β=hyperpolarizability, μ=ground state dipole moment) values, ranging 10-60×10⁻³⁰ esu (electrostatic unit). [42]
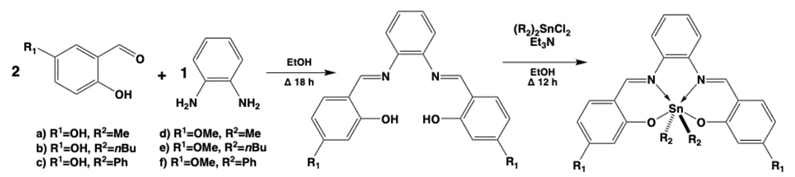
Fig. 8 Two step synthetic methodology to obtain the high π-conjugated hexacoordinated diorganotin(IV) compounds.
Some years ago, our colleagues, José María Rivera, et al. reported a series of 10 new chiral complexes of (salicylaldiminate)tin(IV) Schiff bases, from the Schiff base condensation of 4-(diethylamine)salicylaldehyde and several amino acids in the presence of diphenyltin(IV) oxide. Various of these compounds crystallize in the chiral orthorhombic space group P212121 and investigated their quadratic NLO properties, Fig. 9. At the molecular level, these coordination compounds possess similar electronic spectra (λmax≈ 395 nm) and thus molecular hyperpolarizabilities (β), relative to the same “push-pull” π-conjugated electronic structure. In the solid state, they exhibited efficiencies for second harmonic generation (SHG) up to 8 times greater than urea. The SHG intensities of some of these chromophores seem to be highly correlated with a simple parameter of “degree of chirality” (dχ), defined from the molecular geometries. The working group left a possible quantification of chirality as a perspective. [54]
Also, our colleagues Víctor M. Jiménez-Pérez, et al. developed organotin(IV) Schiff bases for being applied in fluorescent bioimaging (Fig. 10), because it is an excellent tool in cellular biology, and it will be a powerful technique to make out cancer cells. [55]Where intracellular pH value is an important difference between normal and cancer cells. Therefore, the development pH-responsive fluorescent materials are required. Organotin(IV) Schiff bases developed halofluorochromic behavior in solution. Microwave-assisted synthesis achieved better reaction times and chemical yields compared with conventional heating. These compounds were characterized by spectroscopic and spectrometric techniques. Halofluorochromism study evidenced how some molecules in acidic media have the maximum luminescence intensity due to protonation of the organometallic coordination compound. All the fluorescent tin complexes driven, showed cell staining on hepatocyte and MCF-7 cells by confocal microscopy. The theoretical DFT study enabled them to rationalize the optical properties and the halofluorochromism for two compounds synthesized in this work. Their results have shown that the emission decrease, in the acid and basic media for the first and second compounds, respectively, and presumably this is caused by an intramolecular charge transfer (ICT) deactivation process.
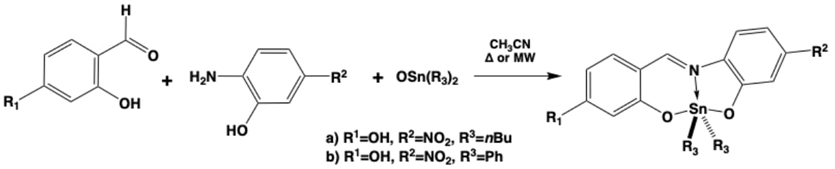
Fig. 10 Synthetic strategy to yield salicyliden and ortho-amino-phenol substituted diorganotin(IV) coordination compounds as halofluorochromic dyes with bioimaging capacities.
Again, our colleagues synthesized a group of diorganotin(IV) coordination compounds with hydrazone-based Schiff bases to apply them in the detection and visualization of cancer cells. The team showed that two of the compounds have mono-photonic fluorescence, but also exhibited efficient bi-photonic excitation. Two of the other compounds, which have hydroxyl substituents, could selectively accumulate in HeLa cells, allowing for the differentiation of normal cells (periodontal ligament cells). Compounds 1 and 3 (Fig. 11) showed excellent staining results of cancer cells (HeLa) using two-photon bioimaging, which is promising not only for bioimaging purposes but for other biomedical applications, and even microsurgery. [56]
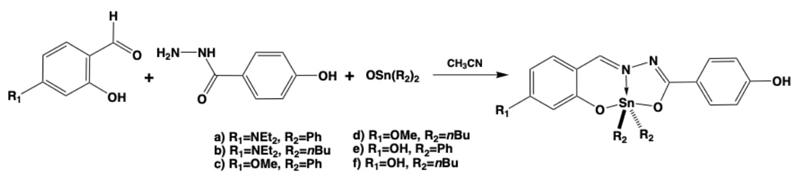
Fig. 11 Diorganotin(IV) coordination compounds with hydrazone-based Schiff bases for the detection and visualization of cancer cells via two-photon absorption process.
Another work of our colleagues performed the multicomponent reaction of 2-hydroxynaphthaldehyde with 2-aminophenol derivatives and its in-situ coordination with diphenyltin(IV) oxide, resulting in the 2-hydroxy-naphythylidene Schiff bases tin(IV) coordination compounds, where the tin atom is pentacoordinated with distorted trigonal bipyramidal (TBP) geometry. [57]They also isolated the 2-hydroxy-naphythylidene Schiff bases ligands. The structural characterization of these compounds revealed that the π-conjugated vertebral column is essentially flat, and the metallic center fixated and ascertained this flatness, and as stated presented a TBP geometry. The fluorescence spectroscopy of both free imines and the organotin(IV) coordination compounds was determined in solution, showing quantum yields of about 4 % with lifetimes between 10−10−10−11 s. They showed a preliminary study as their use as organic light emitting diodes (OLEDs) with a spun film of a nitro-substituted analogue; the device showed intrinsic electroluminescent properties with a threshold voltage of 13 V, a high current density (500 mA·cm2) but a quite low luminance capacity (0.03 cd·m2).
Some of our publications have attracted the attention of Dr. Sedaghat’s research group aiding each other to enhance the organotin(IV) chemistry and possible biological applications. Therefore, her group further studied and applied the gathered knowledge in some points of her research work. For instance, new bis-diorganotin(IV)-based compounds Me₄Sn₂L, Ph₄Sn₂L and Bu₄Sn₂L were synthesized and characterized, and employed the 2,2′‐bis‐substituted diphenylamine arοylidene hydrazone ligand (H₄L) and the coordination environments of both tin centers are pentacoordinated. The L molecules adopt E‐configuration and keto‐tautomeric form in the solid state. In every coordination compound, the bis-hydrazone acts as a tetra-anionic ligand with two contiguous ONO tridentate domains providing a double bis-chelate backbone able to efficiently coordinate the two R2Sn moieties in the enolate form. DNA‐binding studies of the title compounds were performed by UV-vis spectroscopy, evidencing that this coordination compounds significantly interact with calf thymus‐DNA in an intercalative mode. This workgroup also developed polymerase chain reaction assays revealing that all the coordination compounds affected DNA amplification, and that diorganotin(IV) coordination compounds are more effective than the free ligands. The in vitro cytotoxicity of these diorganotin(IV) compounds against the human breast cancer line (MCF7) was determined, indicating that H4L ligand and the dibutyltin(IV) compound developed higher activities in the 25-100 μM concentration range.[58]
Coordination polymers and metal-organic-frameworks (MOFs)
A coordination polymer is a coordination compound in which the monomeric moieties are periodically repeating and interacting each other by coordinative bonds, this means that one monomer is coordinating the other to self-assemble the polymeric material. This building is able to develop in 1D, 2D, or 3D structures with coordinative bonding occurring in these same 1D, 2D or 3D dimensions. On the other hand, MOFs are a subclass of 2D or 3D coordination polymers whose main distinction against the normal coordination polymers is the presence of permanent porosity associated with the generation of voids or free spaces by the self-assembly and rigid directionality of the linkers and metallic ions or clusters.[25,28,35]
The workgroup has been designing and developing coordination polymers since the early 2008 and precisely MOFs of cadmium, which resulted doubly pillared, enabling pore size tunning.[59] Therefore the reaction of the potassium salts of 1,2, 1,3 or 1,4-trans-cyclohexanedicarboxylic acids (trans-chdc), 4,4’-bipyridine (4,4-bpy), with cadmium(II) nitrate in ethanol/water mixtures provided dinuclear cadmium clusters [Cd2(μ-O)2O6N4], that resulted ideal building blocks or secondary building units (SBUs) for the generation of three new double-pillared 2D and 3D open-frameworks. The X-ray structure of one of them resulted [[[Cd2(trans-1,2-a,a-chdc)(trans-1,2-e,e-chdcH)2(4,4’-bpy)2]•8H2O]n] showing axial-axial and equatorial-equatorial conformations at the trans-chdc ligands. With this strategy, the new pillared MOFs were obtained, and studied, but resulted quite unstable compared to other carboxylic derived ligands with aromatic, polyunsaturated or other types of rigid motifs, are included in the structure usually employed for MOF synthesis. This is probably due to the high fluxionality of the cyclohexanedicarboxylate ligands. Nevertheless, the simultaneous employment of carboxylate ligands and doubly substituted pillars resulted in a novel structural concept of MOF, that with some modifications will probably allow for pore-size tuning, thus stimulating a more deep and systematic exploitation of this novel structural concept of “reinforced pillars” to enhance their potential.
Lately, during 2010-2013 the development of new strategic synthetic methodologies was assessed to yield MOFs like [Cu3(BTC)2]n (commonly known as MOF-199 or HKUST-1) with modulable physicochemical and electrochemical properties. Therefore, the employment of ultrasound (US), room temperature (RT) stirring, and solvothermal (ST) energy supply strategies in conjoint with free benzene-tri-caboxylate (BTC, also known as trimesic acid “TMA”) ligand and the nitrate copper(II) hydrated salt conducted to very good results, obtaining the HKUST-1 material almost in any of the tested conditions, with good results in the surface area properties and reaction yields. By XRD analysis, it was possible to identify the presence of Cu2O in the solvothermal methodology, revealing quite harsh conditions. These materials were electrochemically studied by cyclic voltammetry (CV), chronoamperometry (CA) and electrochemical impedance spectroscopy (EIS) and obtained results revealed that the resistance to the faradic process increases as follows US, RT and ST, again associated with the presence of Cu2O obtained in the ST approach.[60]
Later, another variant of the synthetic strategy was assessed now with a base activated (3 eq. of NaHCO3) BTC ligand approach, in this case resulted a metathesis “double-displacement” reaction yielding first the sodium salt of BTC ligand, that by instantaneous contact with the nitrate copper(II) hydrated salt conducted to the HKUST-1 material. To determine if the basic nature of the metathesis reaction conducted to new physicochemical properties to HKUST-1, the monitored variables were: a) energy source either from i) US, ii) stirring at room temperature (SRT) or iii) ST, b) solvent in reaction, and c) reaction time. The characterization techniques indicated that in eight of ten synthetic entries the HKUST-1 material was obtained. These materials also presented low-pressure hydrogen uptake as high as 2.44 wt%. And many of them possess high surface areas. Moreover, according to TGA, DSC and a thermal desorption gas chromatography-mass spectrometry (GC-MS) coupled techniques the reaction conditions afforded materials with modulable degrees of Cu (un)coordinated solvents, therefore there were obtained materials with, more or less, chemisorbed H2O molecules. And precisely the material with less Cu(OH2 contents provided better H2 adsorption capability, see Fig. 12. [61]
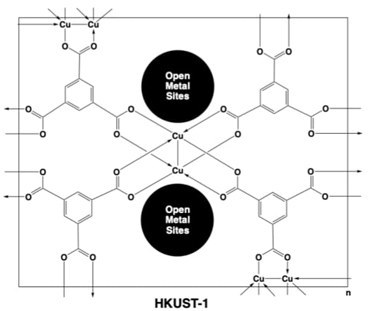
Fig. 12 Structural representative of MOF HKUST-1 with open metal sites provoked by synthetic conditions.
Later, a new ligand based on bis-imide-pyromellitic cores with N-substituted L-alanine was prepared and named (PBIA) and studied its coordination features towards Pr/Eu/Tb/Er/Tm lanthanide metals. By this interaction five isostructural 2D lanthanide (Ln) MOFs were synthesized in DMF leading to LN-PBIA (LN=Pr, Eu, Tb, & Tm) materials (see Fig. 13) [62]. New MOFs were thoroughly characterized where the chemical structures were determined by FTIR, single crystal and powder X-ray diffraction (SCXRD/PXRD) studies, photoluminescence, and scanning electron microscopy (SEM) revealed interesting optical and microstructural features. The PBIA ligands aided the assembly of dinuclear LN-SBUs through monodentante, and (an)iso-bidentate coordination modes of the carboxylate fragment to the LN metallic centers. Some other coordination sites were occupied by the DMF solvent molecules taken from the synthetic conditions, obtaining nonacoordinated (trigonal prism, square-face tricapped “TPRS-9”) centers for the bigger LN and octacoordinated (trigonal prism, square-face bicapped “TPRS-8”) centers for the smaller LN, revealing an important lanthanide contraction effect. Also, in these 2D structures the PBIA ligand plays an important structural role due to its partial mobility/rigidity, coordinating the LN-SBUs in cis (syn) and trans (anti) conformations, being the cis-PBIA-structure assembling and inter-linking 1D coordination polymers, and the trans-PBIA-structure intra-linking 1D polymers to each other to assemble the 2D-sheet. These materials were thermally stable at ca. 400 °C, being activated by a partial removal of the less strongly coordinated DMF→Ln molecules giving PBIA−PrACT/PBIA−EuACT/ PBIA−TbACT/PBIA−ErACT/ PBIA−TmACT phases. This diminishment in coordination number reorganized the monocoordinated PBIA ligands toward isobidentate mode, releasing ca. two of six- coordinated DMF per SBU. The (non)activated Eu/Tb-PBIA materials could be electronically studied in the 200−800 nm range showing characteristic emission bands, revealing that despite of the aromatic nature of the PBIA ligand, it resulted not sensitization-suitable for other Ln emissions. For comparison purposes between (non)activated materials, a band shape modification effect and emission intensity enhancement of 6.7(Eu) and 30.6(Tb) resulted for the Eu/Tb-activated materials. Excitation of PBIA−Eu/PBIA−EuACT directly in Eu3+ resulted efficient, but this was not through the PBIA ligand, and hence a simple antenna effect was not observed. In PBIA−Tb/PBIA−TbACT materials, the PBIA ligand excitation resulted more efficient and PBIA−TbACT compound could be directly excited in Tb3+. An important conclusion herein is that the nonradiative processes are playing decisive roles in luminescent properties. According to the physicochemical and structural characterization of these LN-coordination polymers, isostructural materials resulted, but different LN-metals provided different morphologies/sizes, thus modulating at the microstructural level, and modulating the luminescence properties. These materials would be suitable candidates for molecular/ionic sensing, through emission modulation, mainly when Eu and Tb are employed as the LN centers.
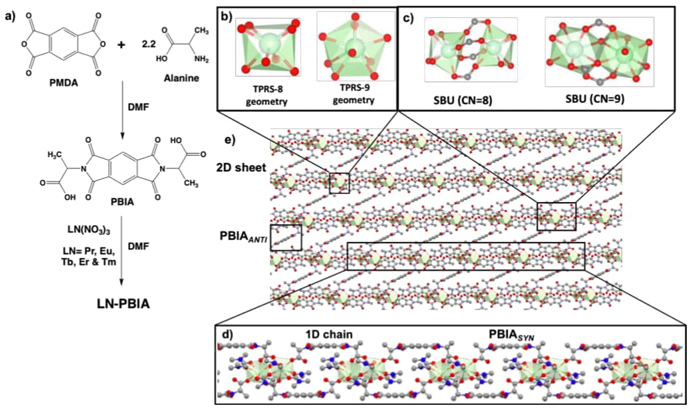
Fig. 13 (a) synthesis of LN-PBIA (LN=Pr, Eu, Tb, Er, & Tm) 2D coordination polymeric materials; (b) coordination polyhedron around small (TPRS-8) and big (TPRS-9) LN centers; (c) dinuclear SBUs in the resulting materials showing the (an)isobidentate coordination mode of four of the six PBIA ligands; d) PBIASYN intralinked-1D coordination polymer; (e) PBIAANTI interlinked-2D coordination polymer.
A derived research work has been obtained by varying the initial reaction stoichiometries between the already obtained PBIA ligand and the LN metal salt, and this initial variation modulated the dimensionality of the resulting material between 2D and 1D [63]. In this case, two new 1D metal-organic frameworks, PBIA-Tm 1D [Tm2(PBIA)2·2(NO3)·4DMF] and PBIA-Er 1D [Er2(PBIA)2·2(NO3)·4DMF], were synthesized from this already known semi-flexible PBIA ligand in conjoint with LN(NO3)3·xH2O salts (LN = Tm and Er) in a 1:1 stoichiometry ratio in DMF. As we already stated in the original contribution, the reaction conditions were carefully analyzed to obtain the 1D structure, and not again the 2D structure previously reported by our workgroup.[62] The two new materials were thoroughly characterized. The SC-XRD structure of PBIA-Tm 1D indicated the formation of a 1D coordination polymer constructed with dinuclear SBUs with PBIA ligands acting as syn-bridging units as in the related 2D-coordination polymers. Each LN center is octacoordinated, with a TPRS-8 geometry, that is linked four times with COO groups from PBIA monocoordinated, and as (an)isobidentate coordination modes, and with one bidentate NO3 -1 (carried in the initial Ln(NO3)3•xH2O salts) and two coordinated DMF molecules placed in the potential voids of the material. Besides, the structure of PBIA-Er 1D was obtained through DFT calculations and Rietveld refinement, taking the PBIA-Tm 1D SC-XRD structure as the starting point due to the similarities observed in their experimental powder diffraction patterns, therefore the LN replacement was carried on and both the cell dimensions and the structural contents were DFT optimized to generate the desired structural model and develop the Rietveld crystallographic refinement to finally acquaint the structure of this new coordination polymer. It was found that the dimensionality of the structure is modulated in the case of Tm and Er materials through the ligand-to-metal ratio/proportion, evidencing that when a 1:1 (ligand:metal) ratio is employed, a 1D material was generated. And when a 2:1 (ligand:metal) ratio/proportion is elected, a 2D material was assembled. On the other hand, this ratio-dimensionality modulation is not viable when lighter lanthanides, such as Pr, are used. Therefore, this modulation in the structure is closely related to both physicochemical characteristics: (i) the lanthanide contraction and (ii) the ligand-to-metal ratio, very promising for dimensionality modulation of materials and thus for further applications. Finally, the TGA of 1D and 2D systems revealed that both have good thermal stability, reaching ca. 400 ºC without degradation of the material.
Infiltrated coordination polymers and MOFs
Another remarkable subject has been encountered in the materials sciences field due to the employment of coordination polymers and MOFs as adsorption materials. This has also been studied in the workgroup with the HKUST-1 compound. Our research group reported in 2016 the encapsulation of the dyes basic fuchsin (BF) crystal violet (CV) and Eriochrome Black T (EBT), which are pollutants present in water sources representing a potential risk for public health and the environment. In addition to demonstrating the efficacy of HKUST-1 in removing this type of pollutants from water, the interaction of the host material with molecules with different electric charges and sizes was studied. In this way, BF, CV, and EBT present a neutral, cationic, and anionic charge, with molecular diameters of 12.40,15.10, or 15.50 Å, respectively (Fig. 14). The encapsulation of the dyes was studied through a post-synthetic methodology (PS) and by a one-pot approach with the dyes in the organic solution (OPO) and in the metallic solution (OPM). In all cases, the encapsulation of the studied dyes was higher than 85 %, representing an outstanding infiltration or loading degree; the best result was observed for the BF-PS experiment and was attributed to the small size of the BF molecule. In contrast, the lowest infiltration degree was observed in the EBT-PS experiment, presumably because to a low diffusion of the dye due to its larger size. XRD demonstrated that the infiltration of dyes did not modify the crystal structure of HKUST-1 but reduced the crystallite size in comparison with the pristine material, thus modifying the microstructural properties of the obtained composites. On the other hand, BET experiments indicated that the dyes@HKUST-1 materials presented a lower surface area compared to the unmodified HKUST-1, corroborating the infiltration of dyes in the cavities of the host material. Finally, TGA revealed that dyes@HKUST-1 presented a decrement in thermal stability in comparison to the pristine material.[64]
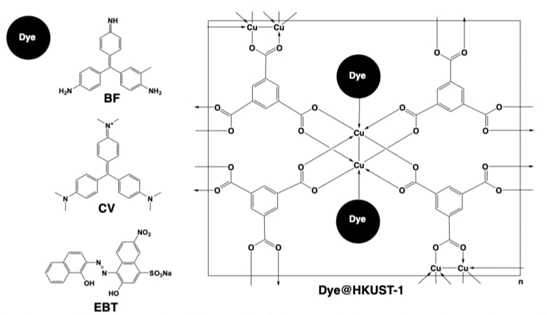
Fig. 14 Loading of BF (neutral), CV (cationic), and EBT (anionic) dyes into the cavities of HKUST-1 material, to obtain Dye@HKUST-1 composites.
Later in 2019, our research group studied the encapsulation of anthraquinone dyes with the purpose of understanding the fundamental interactions conducting the loading of the anthraquinone dyes into HKUST-1 (Fig. 15). The chosen dyes were alizarin (A), alizarin S (AS), disperse blue 1 (B1), disperse blue 3 (B3), disperse blue 56 (B56), and purpurin (P). The encapsulation methodologies conducted for this research were the post-synthetic infiltration, as well as the one-pot methodologies with the guest molecule in the organic solution, and in the metallic solution. The one-pot methodology was more efficient than the post-synthetic alternative since, in the former method, infiltration degrees of 100 % were obtained. The structural characterization of the composites indicated that the structure of HKUST-1 is retained after the infiltration process, except for P infiltrated from the organic solution, in which the linear coordination polymer CuBTC•3H2O is produced. Regarding the stability of the composites, TGA indicated that the degradation range of the infiltrated materials increased in comparison with the pristine material indicating a higher stability associated with the additional supramolecular interaction promoted by the guest molecules towards the MOF. On the other hand, it is worth mentioning that the synthetic process and the guest molecules produce important modifications in the microstructure of HKUST-1 since different morphologies and particle sizes were observed through SEM analysis. Finally, the previous experiments were complemented with computational studies, which corroborated that the lattice parameters of HKUST-1 are not significantly modified by the infiltration of anthraquinone dyes. The optimized geometries also demonstrated that the main interactions between HKUST-1 and the guest are coordination bond type C=Odye→CuHKUST-1, HOdye→CuHKUST-1, and hydrogen bonds type O-HDye⋯OBTC/Ar-HBTC⋯Odye. [65]
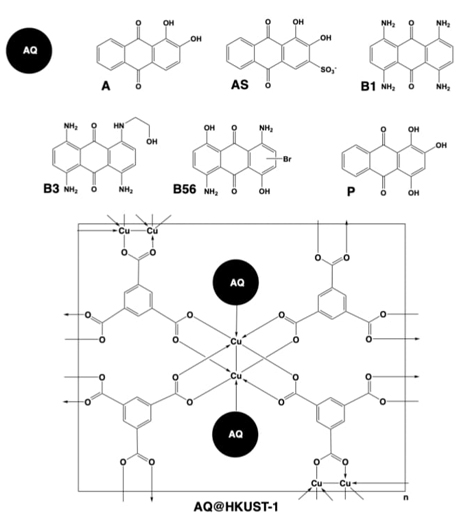
Fig. 15 Loading of anthraquinone (AQ) dyes alizarin (A), alizarin S (AS), disperse blue 1 (B1), disperse blue 3 (B3), disperse blue 56 (B56), and purpurin (P) into the cavities of HKUST-1 material to obtain AQ@HKUST-1 composites.
A different environmental problem than the contamination of water sources by dyes is the presence of greenhouse gases in the atmosphere, such as CO2, which is the main cause of climate change. Consequently, following an environmental approach, the workgroup developed the SWCNT5wt%@HKUST-1 composite (SWCNT: single-walled carbon nanotubes) to enhance the adsorption properties of HKUST-1. SWCNT was employed as a template for the growing of the HKUST-1 crystal on the surface of the nanotubes. The SWCNT@HKUST-1 composite showed an increment in the surface area and pore volume in comparison with the pristine HKUST-1. It is worth mentioning that a mixture of HKUST-1 and SWCNT did not present an increment in the surface area; consequently, the chemical deposition is responsible for the higher surface area. The composite presented a higher CO2 uptake (8.75 mmol·g-1) than bare HKUST-1 (7.20 mmol·g-1) at 196 K up to 100 kPa, we attributed this result to the increment in the surface area. At the same time, the composite showed a higher adsorption heat than bare HKUST-1, 39.1 vs 30.0 KJ· mol-1, respectively. Finally, in-situ Raman spectroscopy was employed to study the adsorption sites of CO2 in the composite and also indicated an increment in hydrophobicity. [66]
Finally, in 2024 we published a review that collects important concepts about the encapsulation of guest molecules in MOFs. In general, there are two strategies to achieve the infiltration of guest species into MOFs: the one-pot method and post-synthetic methodologies. In the one-pot methodology, the encapsulation of guest molecules occurs at the same time as the assembly of organic and inorganic components to achieve the reticular coordination polymer occurred, see Fig. 16. The advantage of this methodology, compared to the post-synthetic alternative, is that the former allows a higher infiltration degree, homogeneity, infiltration of larger species, and infiltration in most internal cavities of the host material. However, in the one-pot the assembly guest species also are able to compete with the organic linkers, sometimes resulting in the formation of crystalline phases different from the host material, being an important drawback. [67]
The second alternative, the post-synthetic method, is useful for the encapsulation of guest molecules with low stability toward the synthetic conditions to generate the host material, since the synthesis of several MOFs requires harsh solvothermal conditions, for example, high pressure, high temperature, high acidic or basic media, see Fig. 16. The post-synthetic methodologies could be classified into two groups: i) infiltration from a vapor phase and ii) infiltration from a solution. In the first approach, the guest molecules must be volatilized, and the host material catches the resultant vapor. In the second methodology, the guest molecules are dissolved in a solvent producing a solution in which the host material is added. The infiltration from a vapor phase is more effective than the infiltration from a solution since, in the former approach, the solvation effect is diminished, as well as the competition between the solvent and the guest molecules for the occupancy of the cavities in the host material. Additionally, the infiltration from a solution usually produces the agglomeration and therefore the self-assembly of guest molecules themselves and on the surface of the guest material, resulting in a lower infiltration degree and diminished homogeneity. For some species, the solubility also could be a problem. In contrast, the main limitation of the encapsulation from a vapor phase is the volatility of the guest molecules, the vapor carrying, as well as the equilibrium between molecular volatility vs. coordination or interaction of the guest molecules towards the MOF. In general, the post-synthetic approach presents the following drawbacks:[67]
poor loading of guest species due to the presence of remnant solvents in the host material,
a higher affinity of the guest molecules with the infiltration media (air or solvents) than with the host material,
low diffusion of the guest molecules through the cavities of the host material,
the collapse of the framework during the infiltration process, and
decrement of important properties such as crystallinity in the resulting composite.
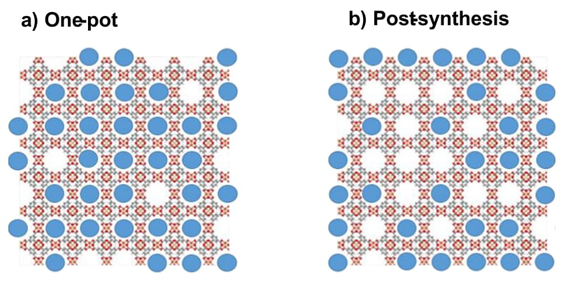
Fig. 16 Depiction of obtained  @material
composites through infiltration strategies: (a) in-situ, while the
material is being constructed the guest species is
present and thus encapsulated in the voids; (b)
post-synthesis, once the material has been already assembled the
guest species is then loaded.
@material
composites through infiltration strategies: (a) in-situ, while the
material is being constructed the guest species is
present and thus encapsulated in the voids; (b)
post-synthesis, once the material has been already assembled the
guest species is then loaded.
Several techniques are useful for confirming the infiltration of guest molecules and the host structure. For example, XRD is an essential tool for confirming the preservation of the host structure through the comparison of the diffractograms obtained from the composites and references, while the shifting of the diffraction peaks and the modifications of crystallinity are related to a successful infiltration. TGA reveals the stability of composites, which usually is altered (enhanced or diminished) due to the presence of encapsulated guest molecules. The observation of new bands attributed to the guest molecules in the FTIR of composites also corroborates the presence of guest species providing insights of the host-guest interaction. Finally, the diminishment of the surface area determined through BET experiments also corroborates the encapsulation of guest species. Regarding the host-guest interactions, they could be determined through FTIR and XPS experiments. In both experiments, the shifting of the bands/peaks indicates modifications in the electronic density of functional groups of the host and guest compounds, which is useful for elucidating the supramolecular interactions. However, the most powerful tool for this purpose are computational experiments due to the optimized geometries of composites directly indicate the most stable host-guest interactions and providing of the resulting chemical structure of the composites, in charge of the obtained physicochemical properties. Nevertheless, these calculations must be supported by experimental evidence. [67]
Bandgap modulation and luminescent properties of coordination polymers and MOFs
Two important properties that have been tracked in the research goals of the group are related to a) bandgap modulation, and b) luminescent properties of studied materials.
Regarding bandgap modulation, in 2024, our research group published a review collecting and discussing the fundamental principles related to bandgap modulation in MOFs and in the resulting guest@MOF composite. The bandgap of non-infiltrated MOFs is usually determined by electronic transition from the linkers, which compose the valence band (VB), toward SBUs, which compose the conduction band (CB). In this way, the bandgap value is directly related to the structure of both linkers and SBUs and the overlap of the p orbital of linkers with the p/d orbitals of metals in the SBUs, where a more effective overlap promotes a bandgap diminishment, hence a lower bandgap. Therefore, the use of elements with frontier orbitals close in energy would enhance the overlap of orbitals, resulting in MOFs with narrower bandgaps. In addition, functional groups in organic linkers could modulate the bandgap of MOFs. Electron-releasing functional groups (-NH2, -OH, CH3, -SH2, among others) increase the conjugation of the electronic density in the organic linkers, enhancing the electronic density itself, and resulting in a partial break of the aromaticity in the system that increases the HOMO energy level. Such an effect produces a VB higher in energy, in comparison with the unfunctionalized material, resulting in a narrower bandgap. In contrast, electron-withdrawing functional groups also increase the conjugation of the electronic system of linkers, but in this case by reducing its electronic density. Consequently, the functionalization of linkers with electron-withdrawing functional groups reduces the energy level of HOMO-LUMO orbitals that also contribute to producing MOFs with narrower bandgaps. However, the mentioned effect occurs only in MOFs in which the band structure is composed mainly of the orbitals of linkers. In the case of halogens, is observed a combination of both effects an increase in the HOMO energy level of linkers due to the donation of lone pairs and a diminishment of the energy of LUMO due to a reduction of electronic density through an electronic-attractive inductive effect. Increasing the conjugation of linkers produces the same effect that halogens since large conjugated systems promote a higher electronic density at the same time that a higher delocalization, which increases the energy of the HOMO orbital and reduces the energy of LUMO orbital, respectively. Finally, functional groups in linkers could also create new energetic states in the band structure of the original material, producing a decrement in the bandgap value. Another strategy to produce MOFs with a narrow bandgap is the creation of supramolecular interactions between the components of the same MOF; indeed, is well documented that MOFs with π stacking interactions between the linkers present narrower bandgaps. [67]
Regarding guest@MOFs systems, we elucidated five mechanisms involved in the modulation of bandgap, see Fig. 17.
I) The most extended and effective mechanism of bandgap modulation is the creation of new energetic states associated with the guest species between the VB and CB of the host materials. Electron-rich guest species usually present HOMO orbitals/VB higher in energy than the VB of the host material, which produces the creation of a VB higher in energy in comparison with the pristine material, reducing the net bandgap value. In contrast, electron-deficient species present LUMO orbitals/CB lower in energy than the CB of the host material, resulting in the creation of a new CB lower in energy that reduces the net bandgap value.
II) The redistribution of the electronic density in the host structure resulted the second mechanism of bandgap modulation. Where electron-releasing guest species promote an increment in the electronic density of the host, increasing the energy of the VB and CB while electron-withdrawing guest species reduce the electronic density of the host and the energy of both the VB and the CB. Thus, to achieve a reduction in the bandgap value a higher increment in the energy of the VB is needed, compared to the CB, for the infiltration of electron-rich species. Whereas for electron-deficient guest species to narrow the bandgap value a higher reduction in the energy of the CB is needed, compared to the reduction in energy of the VB.
III) The creation of a new band structure associated only with the guest molecules was the third mechanism of bandgap modulation. This occurs when the guest molecules present HOMO/VB-LUMO/CB with higher and lower energy values than the VB and CB of the pristine material, respectively. Additionally, the guest@host system that presents this mechanism of bandgap modulation usually has weak host-guest interactions.
IV) The structural modification of the host material due to the guest molecule was the fourth mechanism of bandgap modulation. Where some guest species are still reactive species that could modify the structure of the host, for example, through an oxidation process, resulting in a different chemical environment and as a result a bandgap value modulation. Some guest molecules could also alter the crystal parameters of the host structure, producing contraction or expansion of the crystal lattice, also altering the bandgap value.
V) An increment in the delocalization of electronic density due to strong supramolecular interactions and steric hindrance was the fifth, and the final elucidated mechanism of bandgap modulation. In this mechanism, the higher delocalization promoted by guest molecules reduces the energy level of the CB, while an increment in the energy level of the VB is observed, due to the presence of higher electronic density in the complete system. Finally, is important to mention that strong host-guest interactions promoted a more effective modulation of bandgap, and the modulation of the bandgap is useful for many applications, including photocatalytic processes, conductive materials, and sensors, etc.[67]
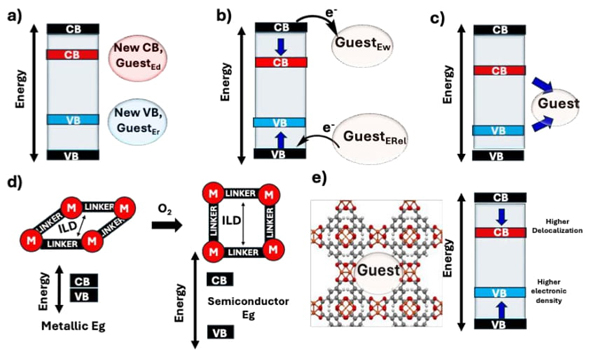
Following the basic principles for modulating bandgap and infiltration, our research group also investigated the effect of remnant solvents (H2O and DMF) on the physicochemical properties of HKUST-1 in 2024. The created materials were named H2O@HKUST-1 and DMF@HKUST-1 according to the solvent infiltrated in the cavities of the host material. XRD and FTIR characterizations confirmed the synthesis of HKUST-1 employing H2O or DMF as solvents. DMF@HKUST-1 presented a higher crystallite size, determined through the Scherrer equation, compared to H2O@HKUST-1, which is a property that enhances charge transport. On the other hand, through FTIR and TGA characterizations, the presence of residual solvents coordinated to open metal sites was observed, while SEM analysis revealed that the solvents employed to synthesize HKUST-1 modify their particle size and morphology. The bandgap value of H2O@HKUST-1 and DMF@HKUST-1 was calculated using the Tauc plots, indicating values of 3.6 and 3.4 eV, respectively. Thus, DMF@HKUST-1 presents better light absorption under the visible region of the electromagnetic spectra, which is a desirable feature for different applications such as photocatalysis. To further understand the modulation of bandgap, computational models were constructed employing periodic DFT calculations; for simplicity and to save computational time, only was considered one infiltrated molecule per cavity of HKUST-1. The optimized geometries for (H2O)1@HKUST-1 and (DMF)1@HKUST-1 indicated that residual solvents are coordinated to the open metal sites of the host material, matching the TGA results. On the other hand, the theoretical bandgap value calculated resulted in 3.6 and 3.3 eV, in agreement with the experimental results. The bandgap decrement was attributed to an increment in the energy of the VB of the host material due to the electron-releasing nature of the guest molecules and to a decrement in the energy of the CB associated with a π-backbonding effect that reduced the electronic density of the SBU. Finally, the materials were evaluated toward a practical application, the photocatalytic hydrogen evolution, in which DMF@HKUST-1 presented the better activity due to its narrower bandgap and higher crystallinity. Nevertheless, both materials showed instability problems related to the formation of the linear coordination polymer [CuBTC•3H₂O].[68]
Our research group recently started analyzing luminescent properties in MOFs, as has already stated in the 2D-LN-PBIA systems. Additionally, in 2022, three MOFs based on Ag as the metallic source and dicarboxylate ligands were synthesized. The workgroup implemented a new methodology that reduces the time of reaction from one week (employing solvothermal or reflux methods) to one day, obtaining moderated yields. The new methodology was applied to the reported Ag2BDC material but also to two new materials, which were named Ag2NDC (UAM-1) and Ag2TDC (UAM-2). It is worth mentioning that the crystal structure of UAM-1 was elucidated by employing a combination of Rietveld refinement and DFT calculations. This represents an important advance for crystallography in Mexico since the number of crystal structures determined through this methodology is scarce compared to the structures determined through the traditional single-crystal XRD method. The crystal structure of UAM-1 showed some similarities to Ag2BDC, in particular in the assembly of the CO2Ag fragment that presents the characteristic argentophillic interactions Ag···Ag present in coordination compounds based on this metal. In addition, FTIR showed bands attributed to Ag-O bonds of the materials around 780-740 cm-1, providing additional evidence supporting the proposed crystal structures. Finally, luminescence studies showed the characteristic emission of Ag2BDC related to ligand-metal or ligand-metal-metal transitions. This emission is possibly due to the short distance between adjacent BDC molecules (aromatic stacking) in the material. In contrast, UAM-1 and UAM-2 presented only emissions in the UV region related mainly to ligand-ligand transitions associated with electronic clouds of the linkers in distances higher than 5 Å. Additional stability studies were conducted, which demonstrated that the materials retained their crystal structure in water for 24 h at 37 ºC, but the crystal lattice collapsed in PBS (phosphate-buffered saline), DMEM (Dulbecco modified Eagle's medium), and DMEM+FBS (fetal bovine serum), that easily produced AgCl, generating materials dissasembly. [69]
In addition to analyzing the luminescent properties of materials, the workgroup has developed materials with potential for practical applications, such as the construction of white light-emitting diodes (WLEDs). For example, in 2023, the research group synthesized the [Eu 2 DPA 3 ] MOF. The structure of this material was elucidated and proposed using FTIR, which evidenced the coordination of the DPA linker with Eu, creating the different moieties CO2Eu, CO2Eu2, and CO2Eu3, as has been observed in other systems also obtained by the workgroup. The picolinic mono- and bis-chelates also were observed, confirming the formation of a polymeric structure. TGA also contributed to elucidating the structure of the [Eu 2 DPA 3 ] MOF, indicating a 2:1 stoichiometry for the metal:linker, a dehydrated structure, and a higher stability of the polymer in comparison to the DPA linker. The developed material presented a quantum efficiency of 48 % in a wavelength of 300 nm and 42 % at 395 nm, with an absolute quantum yield of 55 and 31% in the mentioned wavelengths, respectively. The lifetime determined for this [Eu 2 DPA 3 ] MOF was 2.2 and 1.9 ms at 300 and 390 nm. It is remarkable that the emission of the material excited at 300 and 395 nm was practically the same, and a high red-color purity of 98 % was obtained with chromatic coordinates of 0.65 and 0.35. The emission response of the [Eu 2 DPA 3 ] MOF was attributed to an energy transfer from the ligand to metal and direct excitation of Eu3+. Finally, the analysis of temperature-dependent luminescence properties indicated a stable luminescent response in the range of 30-150 ºC, with a decrease in the response equal to 3.5 %, and extinction energy (ΔE) of 0.21 eV. [70]
Biological properties of coordination polymers and MOFs
As emphasized in the last section, one of the most important research goals is that the substances and compounds investigated could be applied in any field. Therefore, coordination polymers and MOF materials were assessed for their plausible biological applications, and these examples are presented below.
After gaining a solid knowledge of the HKUST-1 MOF, we were able to evaluate the fungicidal effects causing growth inhibition of Aspergillus niger, Fusarium solani, and Penicillium chrysogenum using this compound and two variants: copper-doped HKUST-1 NP, oxide nanoparticles (CuO-NP), and sterilized HKUST-1 (which produces the one-dimensional Cu(BTC)(OH)(H2O) coordination polymer structure). The materials were synthesized by magnetic stirring at room temperature under atmospheric pressure and compared with commercial CuO-NP to evaluate toxicity using a radial inhibition growth technique (inhibition halo). The MOFs were fully dispersed in culture media and the fungi were grown on potato dextrose agar plates containing different concentrations of the tested materials. None of the tested materials inhibited A. niger growth, proving to be resistant due to its Cooper-Leach ability. Meanwhile, the use of concentrations between 750 and 1000 ppm against F. solani resulted in complete growth inhibition. In P. chrysogenum, the dose of 1000 ppm led to an increase in inhibition. These results offer a new approach to MOFs for this type of biological application.[71]
Another approach for biological applications of MOFs is their microbial inhibition, preventing colonization and thus banning the formation of biofilms, since these affections play an important role in 75 % of human infectious diseases. It is known that pathogenic biofilms could survive high doses of antibiotics, leading to ineffective treatments and subsequently increased infection rates. For all these reasons, the use of a MOF as an antibacterial coating is a promising strategy. In this line, silver-based AgBDC MOF showed antifouling properties able to suppress not only bacterial adhesion but also potential surface contamination. By confirming the stability of AgBDC (colloidal, structural and chemical) under bacterial culture conditions, its biocidal activity against E. coli, one of the main indicators of resistant Gram-negative bacteria, was demonstrated. Furthermore, its antifouling and biocidal properties were evaluated by a combination of complementary methods such as colony counting, optical density measurement and scanning confocal microscopy when this MOF was molded as an homogeneous thin film.[72]
Another biological application investigated with HKUST-1 was the study of seed germination and the effects of root and shoot growth in the presence of the tested materials. These effects were also analyzed in the linear polymer([Cu2(OH)(BTC)(H2O)]n·2nH2O) obtained from HKUST-1 hydrolysis. These effects were investigated for seven species of higher plants: sweet corn (Zea mays L.), black bean (Phaseolus vulgaris L.), tomato (Solanum lycopersicum L.), lettuce (Lactuca sativa L.), celosia (Celosia argentea L.), Aztec marigold (Tagetes erecta L.) and gypsophila (Gypsophila paniculata L.). The MOF concentrations studied were 10, 100, 500 or 1000 mg·L-1, and the percentage of germination and growth was increased in most species. All these results are supported by the ability of the MOF to adsorb water and provide micronutrients such as C, O and Cu, which serve as a reserve for the plant. The growth of the bud system is more pronounced with HKUST-1 than with the control and the linear polymer, which should be due to the 3D structure that could absorb a large amount of water. All the species tested proved to be tolerant not only to Cu released from the material, but also to the Cu structured in the MOF, at high concentrations compared to many other systems.[73]Finally, it was found that there is no copper fixation, so metallic phytotoxicity was not possible, therefore being promissory results for further studies.
Polyphenol oxidase (PPO) plays a crucial role in the economic impact of the fruit industry due to its role in enzymatic browning. Some MOFs have shown water stability, low toxicity and good adsorption yield, which turning them good candidates for PPO inactivation. Therefore, the Al-based MOFs MIL-53(Al), DUT-5 and MIL-110(Al) were tested as PPO inactivators in apple juice by enzyme-MOF interactions at room temperature, via two possible mechanisms: i) substrate scavengers or ii) modifiers of enzymatic activity. The scavenging behavior of Al-based MOFs was moderate and in the same order of magnitude, with higher adsorption of catechol than that of 4-methylcatechol, both of which were used as model substrates of PPO. The activity of PPO was reduced by MIL-53(Al)/DUT-5 by at least 70 % in 10/30 min, and MIL-110 completeley inactivated PPO in 50 min, giving place to some enzyme-structural changes. Indeed, enzyme-MOF interactions were found to play a central role in PPO inactivation, realizing that these compounds could be employed as an alternative PPO inactivation process, which besides could be easily integrated into juice processing. This latter could be performed maintaining the sensory/nutritional properties of the product, and that the materials could be easily synthesized, even at room temperature, thus being energy cost effective. [74]
Macrocyclic systems and their applications
Also, in the workgroup there were prepared macrocyclic systems, in this case were tin(IV) and silicon(IV) phthalocyanines (Pc) with different substituents attached to both tin and silicon centers (Fig. 18). This was achieved by the reaction of the already prepared dichlorotin or dichlorosilicon phthalocyanine (PcSnCl2 or PcSiCl2) with the sodium salt of a fatty acid, giving place to the displacement of the chlorine atoms with the carboxylate moieties to yield trans-PcSi(OOCR)2 coordination compounds. For the silicon case,[75] the carboxylates were placed in a trans stereospecific fashion, and the silicon center remained hexacoordinated. But for the tin case.[76, 77] the carboxylates were placed in a cis stereospecific fashion, and the tin center changed from hexacoordinated to presumably hepta- or even octa-coordinated due to the anisobidentate coordination mode adopted by the carboxylate moieties, yielding cis-PcSn(OOCR)2 coordination compounds.[76-79] This extra coordination features also required that the tin center displaced from the mean plane of the Pc ligand, and a concave or domed structure at the Pc ligand was adopted.
The cis-PcSn(OOCR)2 compounds were tested as potential corrosion inhibitors due to the presence of a “naked” Pc ligand face, which presumably would interact with the surface of steel specimens in order to protect them from harsh environmental conditions.[76,77]
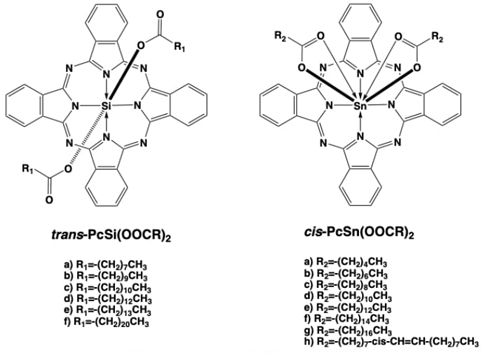
Fig. 18 Chemical structures of trans-PcSi(OOCR)2 and cis-PcSn(OOCR)2 macrocyclic coordination compounds.
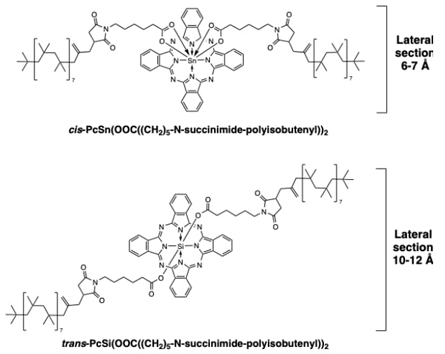
Because of these highly aromatic nature of the Pc macrocyclic core, polymeric substituted derivatives of these silicon and tin systems were molecularly designed, engineered and obtained, to finally assess their potential petroleum asphaltene phase stabilization capability, but also their traceability because of the macrocyclic chromophore core present in these new structures, see Fig. 19. [78, 80] This plausible applicability arose from the knowledge in the oil industry that the asphaltenes in petroleum asphaltenes are the most polar and heavy fraction present in crude oil. Therefore, these complex aromatic molecular mixtures are very difficult to handle and maintain stable, since they tend to self-assemble in bigger particles, even micrometric sized. For these reasons, the employment of chemical stabilizers is a common practice in this field, nevertheless the known efficiencies are low due to the difficulty to maintain flocculated-dispersed states of the mixtures, without precipitation. The research permitted to assemble substituted phthalocyanines of tin and silicon, by the linking of carboxylate-N-succinimidyl-functionalized polyisobutylene (PIB) moieties to the metallic centers, as has been employed in the latter examples of phthalocyanines. The inclusion of PIB polymeric chains is because of the intrinsically disordered structure of this polymer and its efficient interaction towards complex component molecular mixture in crude oil currents.[80] Next was to track the stabilizing behavior of these PcSn(PIB)2 and PcSi(PIB)2 coordiantion compounds by experimental and theoretical means, which were determined by two effects: (i) inhibitive, linked to the stabilization of already formed asphaltenes aggregates or flocculations by an electric field effect, and (ii) dispersive, this linked to the ability of the studied molecules to prevent the size increase of flocculation asphaltene aggregate (clustering). Very importantly, the obtained results revealed that PcSn(PIB)2 is a better inhibitor and dispersant than PcSi(PIB)2, because of stereochemical differences, and to have a naked Pc face, that allow straight interaction with asphaltene molecules, a characteristic that is nos fully accomplished with the PcSi(PIB)2 derivative. Also, because of the presence of the Pc ligand, these compounds have shown high light absorption capacities, and for multifunctional purposes, these two surfactants have shown UV-vis traceability above 100 parts per billion in crude oil media. This is a very desirable characteristic aimed to track and secure transportation and general handling of petroleum and its derivatives.
Supramolecular assemblies and their applications
Supramolecular chemistry has gained efforts, and this has been mainly due to the employment of the coordination chemistry concepts for this new area of chemistry. Supramolecular synthesis has been one of the most studied fields of chemistry during the last 30 years.
The workgroup has obtained some supramolecular approaches intended to understand the modulation of certain physicochemical properties, and concomitantly solve petroleum industry problems. The first was a green mechanosynthesis, free solvent approach has been used for preparing a series of supramolecular complexes. Through directional hydrogen bonds and π-cation interactions, the supramolecular assemblies between the cationic surfactant docecyltrimethylammonium chloride (C12TAC) and phenol (PhOH) could be prepared, and this was achieved with different states of aggregation of matter, where employed stoichiometry between reagents plays an essential role. When the stoichiometric ratio between the two compounds was 1:1, a solid product is obtained. But, when the ratio is higher, products are obtained as gels or pastes reaching a liquid or melted state of low viscosity. Also, the structure of such complexes that formed three-dimensional aggregates between C12TAC and PhOH derivatives could explain the different aggregation states for final products, and this may be the key to understand the observed viscosity reduction, and its mechanism.[81]
In a related paper, theoretical and experimental studies were conducted on the performance of the C12TAC in enhancing the oil recovery factor (ORF) of calcite cores impregnated with heavy oil. The interaction energies between C12TAC and representative molecules of the crude oil phase such as an asphaltene (Asph), a resin (Res) or an octanoic acid, between these molecular assemblies and the calcite surface, were theoretically determined within the framework of density functional theory (DFT). [82]Experimental analyzes based on FTIR, and NMR spectra were performed on the heavy crude oil extracted from the calcite cores by spontaneous imbibition tests. The results show that using a brine with a total ion content of 2.9×105 ppm, ORF values of 8.9 % were obtained at a temperature of 90 ºC and atmospheric pressure, and was of 36 % at 150 ºC temperature and 145 psi pressure. The addition of C12TAC at the same temperature and pressure conditions used in the pure brine experiments resulted in significantly increased ORF values of 36 and 44%, respectively. Based on the molecular interaction strengths between C12TAC and the fractions present in the oil, the splitting of the oil and the subsequent reduction in oil viscosity could also be demonstrated, but without any significant change in the wettability of the calcite core. An analysis of the saturated, aromatics, resins and asphaltenes (SARA) components evidenced a significant change in composition, increasing from their original values of 20.6, 21.0, 35.5 and 22.9% to 31.0, 23.2, 28.7 and 17.2 %, respectively. The lack of change in the wettability of C12TAC in this process is due to a weak interaction in the C12TAC:Res and C12TAC:Asph molecular pairs, followed by a significant decrease in the Res and Asph contents in SARA, indicating that Asph and Res cannot be removed from the rock surface with the employment of this surfactant. The supramolecular assembly between the cationic surfactant and the oil molecules is described by analyzing the molecular orbitals analysis trough quantum chemical approaches. This study has a enormous economic as well as environmental significance, since the oil recovery from the already depleted oil wells is of great interest to improve oil recovery and to achieve greater economic benefitsdue to a larger proportion of extraction with this enhanced recovery. And at the same time, this enhanced oil recovery will reduce the environmental impact as the contaminated oil will disappear from the reservoirs because it has already been extracted from the well.
Another interesting, derived research result is the quantum design and synthesis of a gasoline marker based on a phenol-functionalized supramolecular quaternary salt coordination assembly. To this end, we have presented the development of supramolecular surfactants (SSs) based on MR·OA·(PhOH)i supramolecular assemblies between methylene red (MR), oleylamine (OA) and stoichiometric “I” amounts of PhOH. This was developed with the aim of obtaining markers for the identification of gasolines, using their stability and homogeneous distribution in gasoline mediated by supramolecular interactions and their very characteristic NMR, FTIR and UV-vis spectroscopic evidence. DFT in conjunction with the conductor-like screening model (COSMO) have shown that the supramolecular interactions are due to a non-ionic to ionic reaction in which the carboxyl proton is transferred from MR to OA, and that PhOH confers solubility to the SSs in gasoline by masking their ionic heads. Proton transfer was confirmed by 1H and 13C NMR, FTIR and TGA, while solubility in gasoline was confirmed by UV-vis absorption measurements, which together with viscosity measurements also revealed that the maximum content of PhOH for adequate performance is i=3. This in agreement with DFT-COSMO calculations predicting that for i≥3 the interaction energy of the SSs assemblies becomes weaker per additional PhOH. All theoretical and experimental results support the suitability of the MR·OA (PhOH)i supramolecular assemblies as gasoline markers due to their full compatibility with gasoline and their well-defined spectroscopic fingerprints.[83]
Conclusions
Coordination chemistry is a highly versatile and linking research area since coordination compounds could be applied in several fields, such as traditional catalysis, heterogeneous catalysis, photocatalysis, asymmetrical catalysis, optoelectronics, petrochemistry, antibacterial agents, environmental remediation, drug storage-delivery systems, among many others.
Over 22 years, the research group led by Professor Hiram Isaac Beltran Conde has successfully developed several compounds with applicability in all the mentioned areas, where the obtained systems were organic ligands, boron and tin coordination compounds, tin and silicon phthalocyanines, supramolecular assemblies, coordination polymers, and metal-organic frameworks (MOFs).
In the first years of his career, Professor Hiram Isaac Beltran Conde focused on working with organic synthesis to develop organic ligands and obtain molecular coordination compounds with them. From this period is remarkable the development of synthetic strategies following the one-pot approach, that lately other workgroups remarked as multicomponent approaches, like those found in organic synthesis.
Thus, the expertise of the workgroup in the synthesis of known materials and new materials comes from the first years of the career of Professor Hiram Isaac Beltran Conde. Experience in characterization tools such as RMN, DRX, FTIR, TGA, SEM, AFM, UV-vis, Luminescence, ITC, DSC, and DFT tools arises from the whole research period.
More recently, the workgroup has used the constructed knowledge to develop coordination polymers, mainly MOFs. Under this new approach, the workgroup has optimized the synthesis of known MOFs under greener approaches, as is the case of HKUST-1.
Also, the developed materials have been studied in many applications, always employing an approach of structure/property-activity correlations to understand the macroscopic materials from molecular and structural viewpoints.
All this research involved the design and development of a particular molecular or material system with precise physicochemical properties to achieve a goal, to eventually fulfill a function or be functional or even multifunctional for biologic, scientific or industrial purposes as has been surveyed.
As a perspective for future research, the workgroup will use the constructed knowledge over the years to increase the efficiency of established and new systems in two main applications: the generation of green hydrogen through photo-electro-catalytic methodologies, and the development of efficient luminescent materials since these applications are complementary.
Nevertheless, the approach of investigating emergent properties and applications in the studied materials is not discarded.
Additionally, is remarkable the formation of skilled human resources since all these investigations have been conducted in conjoint with several bachelor, master, and PhD students, as well as with other research colleagues.
Therefore, in in our opinion, the research conducted in our workgroup has contributed to the development of several research fields in Mexico, and abroad.
These results have served other research groups, to publish and even patent interesting synthetic strategies and particular usages of their own systems of study, aiding these other research groups to understand their own studied systems and has furthered the research field of applied chemistry at a multifunctional level.
A high number of citations of the published papers by the workgroup and the inspiration of other research groups in our work to establish their research area demonstrate the previous statement.

