











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
In 1937, Linus Pauling defined hydrogen bonds as a cohesive force relevant to the stability of molecules. In this definition, hydrogen bonds present hydroxyl or amino groups with carbonyl or hydroxyl groups considered as receptors [1]. The main idea at that time considered only oxygen and nitrogen atoms as donors or acceptors of the hydrogens that formed the interaction. This kind of interactions are now named conventional hydrogen bonds.
The possibility that other types of atoms might form hydrogen bonds was questioned for a long time. The existence of other hydrogen bonds with different donor and acceptor atoms that are not typically electronegative is currently recognized. This is the case of the carbon atom in the C-H bond to form a C-H•••O interaction. Such interactions are called non-conventional hydrogen bonds.
Although the hydrogen bond in the International Union of Pure and Applied Chemistry (IUPAC) gold book [2] continues to be defined with basically the same characteristics as it was at the beginning, the same organization recognizes a new definition of hydrogen bond [3] that integrates the new discoveries of interactions. A new broad but not vague definition is intended that considers the visions of different areas of our discipline, but it is clear that this is a difficult task. After several discussions, finally in 2009 the following new definition was established [3]:
“The hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X-H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation. The evidence for hydrogen-bond formation may be experimental or theoretical, or ideally, a combination of both. A typical hydrogen bond may be depicted as X-H···Y-Z, where the three dots denote the bond. X-H represents the hydrogen-bond donor. The acceptor may be an atom or an anion Y, or a fragment or a molecule Y-Z, where Y is bonded to Z. In specific cases X and Y can be the same with both X-H and Y-H bonds being equal. In any event, the acceptor is an electron-rich region such as, but not limited to, a lone pair in Y or a π-bonded pair in Y-Z”.
In this definition, different types of atoms can be considered as hydrogen donors or acceptors. It also establishes that it is essential to have evidence. In this sense, theoretical and computational chemistry contributed greatly to the study of hydrogen bonds, not only to provide evidence, but to study their nature, which in many cases have not been completely clarified.
The role of hydrogen bonds (conventional and unconventional) is recognized today as the main intermolecular interaction in various chemical systems and different materials. Other types of intermolecular interactions, where there is still much research to be done, have appeared on the scene [4-8] that may be as important as hydrogen bonds, namely dihydrogen bonds (H•••H bonds) and non-covalent interactions between heteroatoms.
In our group, conventional and unconventional hydrogen bonds as well as other intermolecular interactions, have been studied for more than 20 years using theoretical and computational chemistry tools. This paper summarizes our main contributions to this topic. It is not intended to be a review article, but rather to highlight the work that has been developed in the Department of Chemistry at the UAM Iztapalapa, which has contributed to the understanding of non-covalent interactions in many different systems. The UAM is a recently created University, which is turning fifty years old, but important research work has been carried out in many fields, and Theoretical and Computational Chemistry is not an exception.
Biological macromolecules
Hydrogen bonds are weaker than covalent bonds or interactions between ions, but it has been recognized that they can be crucial to form the structure of biological macromolecules such as proteins, DNA or carbohydrates. Conventional and non-conventional hydrogen bonds can be found in these macromolecules, with a wide range of interaction energies. The interaction energies of hydrogen bonds depend on the atoms involved and on the geometric parameters such as the distance between the acceptor (A) of the bridge and the participating hydrogen (H), the distance between the acceptor and the donor (D) of hydrogen, and the D-H•••A angles. Shorter distances and more linear angles imply higher energies (more strength) of the hydrogen bond. With these parameters, hydrogen bonds can be classified as strong, medium-strength or weak, considering Jeffrey's classification as a reference [9]. This classification is as follows: Jeffrey categorizes H bonds with donor-acceptor distances of 2.2-2.5 Å as “strong, mostly covalent”, 2.5-3.2 Å as “moderate, mostly electrostatic”, 3.2-4.0 Å as “weak, electrostatic”
With this classification, many systems can be studied. As an example, we consider the C-H•••O hydrogen bond that was one of the most controversial and relevant weak interaction in the structure of proteins and carbohydrates. In 2000 [10] our group estimated, by using N,N'-dimethylformamide dimers as a model and high-quality ab-initio calculations, a binding energy of the C-H•••O=C interaction between 2.1 and 4.0 kcal /mol depending on the linearity of the bond. The role of linearity in the strength of C-H•••O=C hydrogen bonds was clearly demonstrated by Density Functional Theory (DFT) calculations [11]. Although each hydrogen bond is weaker than conventional hydrogen bonds, the presence of many C-H•••O bonds strengthens the interactions and contributes to the conformation of biological macromolecules.
Many computational codes use Gaussian functions to represent orbitals. Although such functions are convenient for some numerical approaches, estimations of intermolecular interaction energies exhibit the Basis Set Superposition Error (BSSE). Usually, the estimation of the BSSE is obtained by using the counterpoise method [12-14]. We have shown that this error can be reduced when using Kohn-Sham (KS) method instead of correlated methods based on the wave function [15]. Therefore, if the basis set is large enough, in DFT-KS calculations the BSSE can be negligible on the estimation of hydrogen bond energies.
The new definition of hydrogen bonds suggests that there must be theoretical or experimental evidence to ensure their presence. Theoretical and computational chemistry are useful tools to provide this evidence based on the analysis of the electron density. Scalar and vector fields of electron density are very important to find and characterize intermolecular interactions. To this purpose, Quantum Theory of Atoms in Molecules (QTAIM) [16] has been widely used to establish intra and intermolecular interactions, in particular, hydrogen bonds. The electron localization function (ELF) is another scalar field that is also used for this purpose[17]. By testing conventional and non-conventional hydrogen bonds, our group found that critical points of the electron density and ELF are localized almost in the same contact region giving a relationship between both approaches [18].
Graphics processing units for atoms and molecules
Due to the importance of the analysis of scalar and vector fields of electron density to find and characterize intermolecular interactions, our group developed a software to use Graphics Processing Units (GPUs), and thus enable to study of non-covalent interactions in large molecules. This software has been called GPUAM, which stands for Graphics Processing Units for Atoms and Molecules [19-21]. With this software it is possible to analyze the gradient and the Laplacian of the density, to find the critical points that denote interactions, to obtain other fields such as the ELF, the electrostatic potential, and the Non-Covalent Interactions (NCI) index for large systems [22,23]. The GPUAM has been meticulously designed to run efficiently on GPUs using CUDA-C programming techniques. The grid used to analyze 3D scalar or vector fields is distributed across all threads involved in one or multiple GPUs within a server. Therefore, the efficiency of GPUAM is directly related to the number of threads available in the GPUs. Detailed information about the kernel designed in CUDA for grid distribution can be found in reference [19]. This grid-based approach is well-suited for GPU implementation, making the search for critical points in electron density highly efficient. Consequently, the QTAIM can be applied to large systems, as discussed below.
By using GPUAM, it is possible to investigate models of cavities of up to 800 atoms to study the interaction of drugs with proteins at the level of ab initio DFT methods. As an example, with QTAIM and GPUAM we characterized the non-covalent interactions between the N3 inhibitor and the main protease of SARS-CoV and SARS-CoV-2 [23]. We found that there are conventional and unconventional hydrogen bonds, as expected, but also H•••H interactions that are generally not considered in these systems.
Using the same methodology, we also studied the intermolecular interactions between risperidone and the dopamine receptor DRD2 [24]. Risperidone is a drug used to treat schizophrenia. We compared the interactions of dopamine with DRD2 and those of risperidone with the same receptor, all using the QTAIM and the NCI (Figure 1). We found that the interaction energy depends more on the number of interactions than on the strength of each interaction. In this same topic, we show the importance of the salt bridge in the interaction of antipsychotic drugs with the dopamine receptor and its relationship with reactivity indices generated by DFT [25].
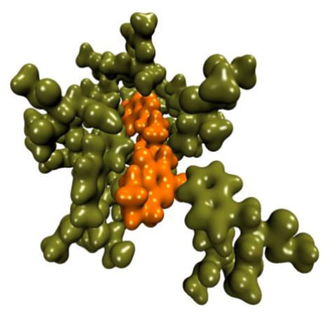
Fig. 1 Electron density in the Risperidone (orange) - DRD2 (green) system, which contains 571 atoms.
We also studied the intrinsic chemical reactivity of several drugs, dopamine agonists and antagonists, and their interaction with DRD2 in order to elucidate the action mechanism. We found a correlation between the ability to donate or accept charge and the strength of the interaction with the dopamine cavity in DRD2 [26-28].
Inclusion complexes
Macromolecules form cavities and this is very useful for housing small molecules without altering their structure. The polarity as well as the functional groups that can form hydrogen bonds or other interactions, promote the inclusion of small molecules in the cavities of macromolecules. Intermolecular interactions between guest and host are the driving forces of inclusion complexes. The formation of inclusion complexes produces favorable changes on physicochemical characteristics of the host, such as solubility, dissolution rate, stability and biodisponibility.
Inclusion compounds with cyclodextrins
Cyclodextrins (CD) are macrocycles with truncated cone cavities that make them good for encapsulating compounds for various applications in the pharmaceutical or food industries. In the inclusion complexes that form CDs, intermolecular interactions between the guest and the host are of great importance. The shape of the CD defines the distribution of the electron density within the cavity. The electron density can be mapped to their electrostatic potential. CDs have negative electrostatic potential in the widest part of the cavity and positive in the narrowest section. Therefore, CDs have a total dipole moment that determines how the guest molecule is introduced and interacts within the cavity. [29].
As we mentioned previously, physicochemical properties of the host are modified by the formation of complex. For example, the optical properties of the included compound. This was demonstrated for 4-dimethyl-aminobenzonitrile. The electrostatic potential of the CD cavity and intermolecular interactions change the fluorescence of the compound. Taking advantage of this experience, 4-dimethyl-aminobenzonitrile was proposed as a CD antenna to be used as an optical sensor [30].
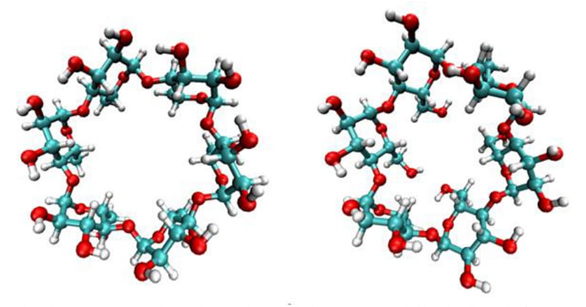
More recently, molecular dynamics calculations show that α, β, and γ-CD cause significant CDs deformations when complexing molecules such as sertraline and abacavir. Ab initio DFT calculations showed that X-ray structures are maxima at the potential energy surface (PES), and that not only hydrogen bonds are responsible for the stabilization of the inclusion complex, but other interactions such as H•••H and Lewis acid base type interactions [31,32]. Deformation of cyclodextrins is an important element to be considered in inclusion compounds. The deformation can be obtained from docking and dynamic molecular techniques, and it is crucial to do this before applying DFT methods to characterize non-covalent interactions. Distortions found in α-CD are presented in Figure 2. Structure on the left side corresponds to that obtained from X-ray experiments. The structure on the right side corresponds to the average structure obtained from classical molecular dynamics. From this figure it is evident the distortion observed when the α-CD is submitted to movements induced by classical molecular dynamics. The adequate description of the PES of different conformations due to conventional and non-conventional hydrogen bonds, depends on the correct description not only of intermolecular interactions, but also intramolecular, as it was demonstrated for the alanine dipeptide [33].
Mesoporous materials
The mesoporous materials such as SBA15(SiO2) have applications in several fields due to the elevated specific surface areas where adsorption of a variety of substances is possible in their channels that can be released under certain conditions. Specifically, we evaluated a material with a SiO2 matrix whose pores trap fluconazole molecules. Fluconazole is a corrosion inhibitor, and the idea is to have a reservoir of a corrosion inhibitor agent to use this material as a self-healing material [34]. From the electron density analysis of structures proposed by docking, we found that the fluconazole is mainly trapped by non-canonical interactions such as F•••O; O•••O; N•••O; C•••O, as well as C-H•••O hydrogen bonds. Just in one structure, conventional hydrogen bonds are observed. The analysis of the nature of intermolecular interactions is important since the liberation rate depends on them. In this study, conditions of release were proposed to use the material in coating metals.
Clathrates
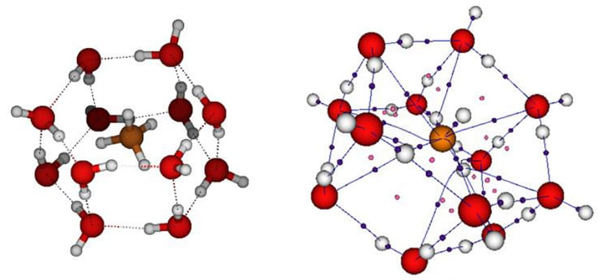
Methane hydrates or clathrates are other compounds that trap molecules such as methane. These are boxes formed with water molecules linked by hydrogen bonds. Non-polar compounds such as methane can be housed inside. Clathrates are found in nature at the bottom of the arctic oceans, and can also be synthesized in the laboratory. They have been thought of as alternative energy sources or as possible structures to store hydrogen. In our group we widely explore the potential energy surface of the formation of the methane hydrate CH4-(H2O)12 with ab initio methods [35]. We found conventional hydrogen bonds between water molecules as expected, and C-H•••O hydrogen bonds when water interacts with methane (Figure 3). The most stable structures found for CH4-(H2O)12 clusters are nest-like instead of cages trapping the methane molecule. Finally, with high quality ab initio methods, we stated that CH4-(H2O)12 clusters could exist up to 179 K, which means that it is possible the existence of these clusters on Mars, that was a discussion topic at that time.
Molecular conformers
Hydrogen bonds and other interactions are also important factors in the structural conformations of different molecules and the physicochemical properties that they can display. For example, hydrogen bonds are crucial for the decomposition of three common neonicotinoids used as insecticides [36] or the formation of clusters of NaCl(H2O)2[37]. The solvation of Cu2+ ion is also important. In this case, we used a modified version of the simulated annealing method to explore the potential energy surface of [Cu(H2O)n]2+ with n=12, 16 and 18 in order to find the coordination number of Cu2+ ion. By using QTAIM we found that the fivefold coordination is preferred and the fourfold coordination cannot be considered in studies where the Cu2+ion is solvated by water molecules [38].
Conformations of N-(2-benzoylphenyl)acetamide [39] are interesting since the most stable conformation in gas phase is stabilized by the hydrogen bond between the N-H of the acetamide and the carbonyl group that joins the two rings. However, another conformer close in energy is stabilized by a weaker C-H•••π. This study makes clear that including dispersion correction in DFT calculations is essential to correctly describe molecular conformations.
Ferulic acid is another interesting example. DFT calculations and electrochemical experiments showed that planar structures play an important role in the antioxidant capacity [40]. Dimers and trimers of this acid show weak intramolecular interactions that produce bent conformers. However, the presence of more than one ferulic acid unit can lead to donating more than one electron. Other example is the adduct formed between 1,4-benzoquinone and benzoic acid that presents a planar configuration, which is stabilized by two hydrogen bond interactions: one involving OH•••O and the other CH•••O. This finding was further supported by electrochemical analyses [41]. The presence of hydrogen bonds is also important in intramolecular contacts, something that was revealed in studies with dopamine [42].
The methodology used gives us the possibility to study Raman spectroscopy signals that allow us to investigate the pathogenesis and progression of Parkinson Disease (PD). It has been reported that 5-S-cysteinyl-dopamine (CysDA) is a toxic compound for dopaminergic neurons, which induces the pathogenesis and progression of PD. To quantify CysDA by Raman spectroscopy, a solid graphene oxide (GO) substrate was proposed in order to increase the intrinsic low sensitivity of the Raman scattering and the interference of the fluorescence signal of biomolecules. We demonstrated that CysDA is adsorbed on GO by a wide variety of intermolecular interactions as hydrogen bonds and interactions between heteroatoms. These interactions explain the main band shifting observed in the FTIR spectrum of the CysDA/GO complex. Furthermore, the interaction of CysDA with GO leads to the quenching of the fluorescence of CysDA, which permits to obtain the Raman spectrum of this molecule [43].
The antioxidant capacity of catechols and resorcinols is well known. Catechol presents an O-H•••O interaction due to the ortho position of the OH in the phenolic ring. When there are different substituents, the energy of the interaction found is modified [44]. The analysis of this interaction with different density fields showed that with the O-H•••O interaction, the O•••O repulsion of the two OH groups is avoided. In both resorcinols and substituted catechols, it was found that the antioxidant capacity is not affected, but “the electron donating groups favor electronic changes along the reaction path, increasing the spontaneity of the hydrogen atom transfer mechanism” [45].
Intermolecular contacts in crystals
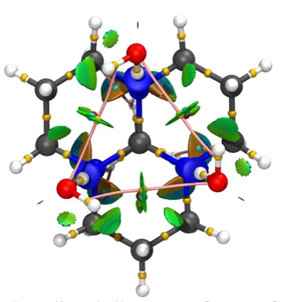
Many-body effects are enhanced in crystals and the behavior of some isolated molecules is altered when they are immersed in a periodic system. For example, tricyclic orthoamides present the eclipsed all-trans conformer more stable than the corresponding alternated conformation, which is quite strange for isolated molecules. The reason for this conformation involves two effects shown in Figure 4, non-covalent interactions with water molecules and the packing involved in the crystal [46].
The packing in a crystal is an important point to be considered for intermolecular interactions since the pressure exerted over a system is mapped on the contacts between molecules. For urea, our group described in detail how the hydrogen bond is altered as a function of the pressure. It is worth noting that the structures used in this study were obtained from experimental data, in particular, X-ray structures [47]. The changes of hydrogen bonds have an impact on the electron density voids observed in the crystal structure as it is shown in Figure 5.
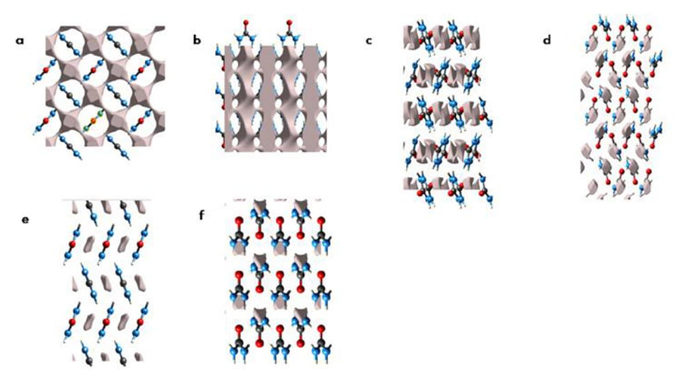
Fig. 5 The gray isosurfaces are the electron density voids of urea crystal at the (001) and (010) planes at 0.0047 GPa (a and b); 1.48 GPa (c and d) and 3.10 GPa (e and f).
For many reasons, the adsorption phenomena are relevant for physical and chemical processes. In these processes, non-covalent interactions are crucial to correctly describe host-guest contacts. In particular, Metal Organic Frameworks (MOF) have been used to confine substances considered as pollutants. Our group investigated systems where MOFs are the host and CO, CO2, SO2, benzene, toluene and I2 are the guests. The MIL-3(Al)-TDC MOF increases its adsorption capacity if molecules of water are within the MOF. This result sounds strange. However, the directionality of hydrogen bonds between MOF and water induces a better interaction between MOF and CO2. This conclusion was obtained from theoretical methods [48]. A similar effect is observed for the adsorption of CO2 in InOF-1 MOF, which is enhanced when 2-propanol is confined within the MOF [45]. The relevance of the InOF-1 MOF to trap substances is evident from experimental and theoretical studies [49-53].
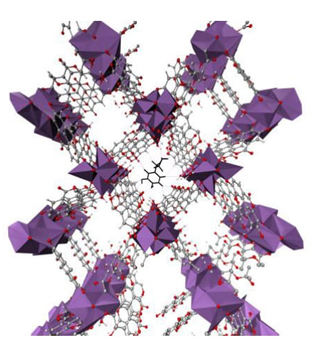
Electronic structure methods are mandatory to elucidate the non-covalent interactions between MOF and guest molecules. CO and SO2 adsorption sites within NOTT-401[54], I2 in MIL-53(Al)-TDC [55] are examples where the analysis of the electron density provides insight of non-covalent interactions in these systems. Another example is the analysis of the electron density and related quantum chemistry scalar fields to understand the differences between isostructural MFM-300(Sc) and MFM-300(In) systems [56].
The MOFs have been considered to be carrier drugs. In our group, dopamine encapsulated by SU-101 MOF [57], and phenylethylamine, dopamine or sertraline within Mg2(olsalazine) MOF [58] have been analyzed as possible drug delivery systems (Figure 6). For both MOFs the analysis of the electron density is quite important, by using NCI and QTAIM analysis we have characterized the difference between the open metal sites in Mg(olz)2 and SU-101 MOFs [58].
Conclusions
In this paper we have mentioned the works where some actual and former professors of the Chemistry Department have contributed to the understanding of non-covalent interaction. From molecules to crystals, intermolecular contacts have been analyzed by using mainly the electron density and related quantum chemistry scalar fields. In all reports mentioned in this paper, the electron density has been obtained through the density functional theory. Our GPUAM software was used in molecular and periodic systems to analyze the electron density scalar fields. Our research has contributed to visualize the important role of non-covalent intermolecular interactions, not only conventional hydrogen bonds, but others as H•••H and heteroatoms contacts. We recognize the effort performed by other groups around the QTAIM and molecular interactions in our country, such efforts have been highlighted in reference [59].
The non-covalent index has been widely explored in our group in all the systems we have studied, however, only qualitative observations can be deduced from here. It is important to have quantitative information from the NCI or other scalar fields, and for this reason we are working on this. More investigations are needed to study other interactions such as those between heteroatoms that appeared in all the systems studied in our group.

