











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Biophysical methods for studying proteins
Studying protein dynamics is not just an academic pursuit. It is a crucial step towards understanding the role of structural flexibility and dynamics in protein activity and catalytic efficiency, which in turn informs the design of proteins with enhanced activity. Differential scanning calorimetry (DSC), fluorescence spectroscopy, circular dichroism (CD) spectroscopy and molecular dynamics (MD) simulation are crucial methods for investigating protein characteristics, providing valuable information on their composition, durability, and performance [1-4]. This combination enables the verification and improvement of theoretical models, resulting in a more comprehensive and precise understanding of the protein folding process.
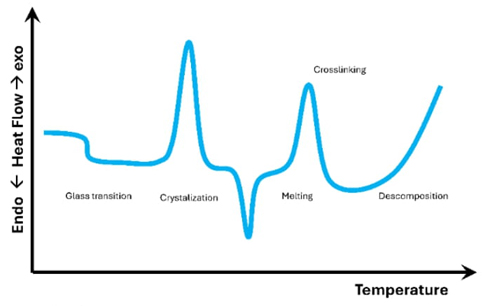
Differential Scanning Calorimetry is an effective method to study how heat affects proteins and how their structures change. It does this by precisely measuring the flow of heat energy caused by changes in structure caused by temperature. This technique provides extensive information on crucial thermodynamic characteristics, such as the melting temperature (Tm), the enthalpy change (∆H), the heat capacity change (∆Cp), the Gibbs free energy change (∆G), and the entropy change (∆S). These measurements are critical for understanding protein stability in a variety of contexts and developing more robust proteins for industrial and therapeutic applications. It is crucial to understand how past processes affect the present behavior of a protein, as in some cases, denaturation processes cannot be reversed, and the scanning rate significantly impacts studies on protein unfolding using differential scanning calorimetry [5-8] Hysteresis, a phenomenon that illustrates the influence of a system's past trajectory on its current state, plays a crucial role in investigating unfolding and refolding processes and comprehending the branching points in folding landscapes [9,10]. Mathematically, the heat capacityCp) of the sample is defined as (equation 1):
where dQ is the amount of heat added and dT is the change in temperature. The temperature difference (dT) between the sample and reference changes each time the sample undergoes an exothermic or endothermic transition, causing a proportional difference in heat flow rate (dQ) which is critical for accurately determining the thermodynamic parameters and understanding the thermal behavior of proteins [11]:
where ∆Cp is the difference in heat capacity between the unfolded and native states, and Ti and Tf are the initial and final temperatures of the transition. The melting temperature (Tm) is identified as the point where the heat capacity exhibits a maximum, indicating the temperature at which the protein unfolds. The Gibbs free energy change (∆G) can be related to ∆H and ∆S using the equation 3 [11]:
Gaining an understanding of these interconnections enables a more sophisticated analysis of differential scanning calorimetry data, which in turn leads to a more profound comprehension of protein thermodynamics and folding processes. This knowledge also aids in the development of resilient proteins for a variety of purposes. A typical plot from a differential scanning calorimetry experiment is shown in Fig. 1. It displays a graph of heat absorbed (endothermic) and heat given off (exothermic) as a function of the sample's temperature. It is crucial to emphasize that Fig. 1 does not represent all possible transitions in a single sample. Rather, it serves as a visual representation of the most often observed forms of transition.
Circular Dichroism is a spectroscopic technique that measures the difference in absorption of left-handed and right-handed circularly polarized light by chiral molecules. This method is widely used to study protein conformation, stability, and ligand-protein interactions. By looking at the different ways that circularly polarized light is absorbed, circular dichroism gives information about the secondary and tertiary structures of biomolecules. This lets scientists figure out how changes caused by mutations, or the environment affect those structures. Ellipticity θ in circular dichroism is related to the difference in absorbance (∆Abs) by the equation 4 [3,12]:
Additionally, molar ellipticity (θm) calculated as (equation 5):
where θ is the ellipticity in degrees, C is the molar concentration of amino acids, and l is the cell length in cm.
The far-ultraviolet spectrum, ranging from 180 to 240 nm, offers a means to assess a protein's secondary structure composition. Fig. 2 shows the characteristic circular dichroism spectra of the secondary structures α-helix, β-sheet, and random coil. In circular dichroism studies of unfolding processes, the spectra can be taken at different temperatures to monitor structural changes; another, more convenient, option is to monitor those changes as the temperature is continuously varied with a constant heating ramp, which allows for the calculation of the fraction of unfolded protein and thus the equilibrium constant for the process. The van’t Hoff analysis, derived from the temperature-dependent equilibrium provides the van't Hoff enthalpy (∆H) and entropy (∆S), the midpoint of the unfolding transition (Tm), and the unfolding free energy (∆G). These parameters are crucial for understanding the stability and folding behavior of proteins under varying thermal conditions [12-14]
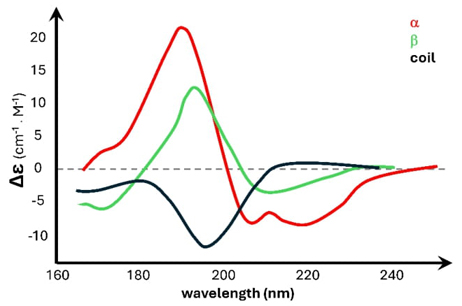
Fig. 2 Displays the circular dichroism curve representing the typical secondary structures: α-helix (red), ꞵ-sheet (green), and random coil (black).
Fluorescence spectroscopy, on the other hand, is a technique used to study the emission of light from a substance after it has absorbed light of a different wavelength. Fluorescence spectroscopy is an analytical method that is both very sensitive and selective. It is used to study the structural and thermodynamic characteristics of proteins. This approach utilizes the fluorescent properties of amino acids like tryptophan and tyrosine to detect proteins at low levels and observe real-time changes in protein structure, such as folding and unfolding. The thermodynamic parameters assessed include the free energy of the denaturation process and its cooperativity. Fluorescence intensity is related to the concentration of the fluorophore by equation 6 [15,16]:
where IF is the fluorescence intensity, k0 is a constant depending on the detection system, ϕF is the fluorescence quantum yield, and [F] is the fluorophore concentration expressed like (1-10-εcl).
If dilute solutions are used so that less than 2 % of the excitation energy is absorbed, an approximation can be made that simplifies the fluorescence spectroscopy analysis. In this scenario, the absorbance (Abs) is sufficiently low, allowing us to assume that the relationship between fluorescence intensity (IF) and the concentration of the fluorophore [F]) is linear. This linearity arises because Beer-Lambert's law, which describes light absorption, can be approximated as follows for very low absorbance values (equation 7) [17,18]:
Protein fluorescence, particularly from tryptophan residues, is highly sensitive to the molecule's surrounding environment. Tryptophan residues inside the protein's interior are found in less water-soluble environments, resulting in a change in the wavelength at which they emit light. In nonpolar settings, the fluorescence maximum occurs at shorter wavelengths, whereas in polar situations, it shifts towards longer wavelengths. So, changes in the protein that affect the area around tryptophan residues, like protein-ligand interactions [19], structural changes, or unfolding, will change the fluorescence spectrum. Fluorescence's sensitivity makes it an excellent tool for examining protein dynamics, as seen in Fig. 3.
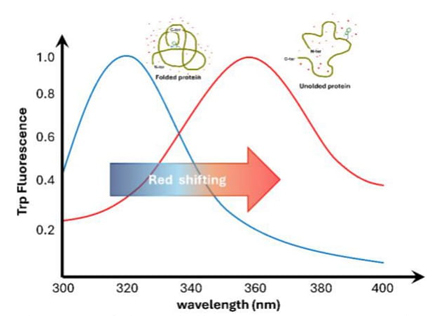
Fig. 3 Intrinsic fluorescence. Protein unfolding, followed by the movement of a tryptophan residue from the hydrophobic internal enviroment, to the aqueous outside (shown by the red dots). The tryptophan fluorescence spectra are shown on a graph under two conditions: native (blue) and denaturied (red). The redshift is found (arrow) when the maximum emission moves toward longer wavelengths.
Molecular Dynamics simulation enables the observation of protein’s dynamic behavior at the atomic scale, providing vital atomistic information to comprehend the fundamental principles of protein stability and unfolding. These simulations have the capability to compute thermodynamic characteristics such as Gibbs free energy (∆G), enthalpy (∆H), entropy (∆S), and, under some circumstances, heat capacity change (∆Cp) [1,18,20,21].
Newton's second law is used in molecular dynamics simulations to calculate the trajectories of atoms (equation 8):
where F is the force acting on an atom, m is the atom's mass, and a is the atom's acceleration.
The interaction law is determined by the potential function U(r1,…,rN), which represents the potential energy of N interacting atoms based on their locations, ri=(xi,yi,zi). The force acting on the i-th atom is dictated by the gradient in relation to atomic displacements. The equation Fi=∇ri U(r1,…,rN) represents the relationship between the force Fi and the position vector ∇ri U [22].
The components of the gradient are given by (equation 9):
Discovering an accurate potential to replicate authentic energy surfaces is complex, yet it simplifies calculations. Atomic force field models and classical molecular dynamics rely on empirical potentials that include specific functional forms to accurately depict the system's physics and chemistry. The following is an example of a force field commonly used in biosystems simulations (equation 10):
In this equation
The first term sums over all bonds, with ai representing the bond strength and (li-li0 ) being the deviation from the equilibrium bond length.
The second term sums over all angles, with bi representing the angle force constant and (θi-θi0) being the deviation from the equilibrium angle.
The third term sums over all torsions, with ci representing the torsional force constant and [1+cos(nωi-γi0)] accounting for the periodic torsional potential.
The fourth term sums over all pairs of atoms not bonded chemically, with
The final term sums over all pairs of point charges qi and qj separated by distance rij, representing the Coulomb potential
Replica exchange molecular dynamics (REMD) is an advanced approach in Molecular Dynamics simulation that greatly enhances the exploration of different conformations in a protein's structure. This technique is particularly useful for studying the intricate mechanisms involved in protein folding [23,24]
The combination of these experimental and computational methodologies allows for the examination of mutation consequences, exploration of protein-ligand interactions, and comprehension of the variables that influence protein aggregation, folding, and unfolding. Adopting this approach is crucial for enhancing our comprehension of basic biological events and has substantial implications in the fields of drug development and biotechnology. Table 1 displays the essential experimental techniques and their corresponding thermodynamic parameters used to evaluate protein stability.
Gaining a comprehensive understanding of the processes involved in protein folding and unfolding is crucial in the field of biophysical chemistry research. This knowledge allows for significant insights into the structure, dynamics, and function of proteins. Understanding this information is vital for proteins like triosephosphate isomerase (TIM), glucosamine-6-phosphate deaminase (G6PD), laccase, and bacteriophage M13, since they perform critical tasks in many biological processes [7,25-33].
The researchers in the Department of Biophysical Chemistry at the Universidad Autónoma Metropolitana (UAM), Iztapalapa campus, are dedicated to understanding the processes involved in protein folding and unfolding. These mechanisms have a profound impact on catalysis, metabolism, and biological activity. Scientists use experimental methods such as spectroscopy and calorimetry to examine the changes in protein structure and stability under various situations, such as fluctuations in temperature and variations in denaturant concentration. The study's findings provide important insights into the molecular pathways responsible for protein misfolding and aggregation, which are occurrences associated with neurodegenerative disorders. For instance, the processes of unfolding and refolding TIM proteins take place gradually within a certain temperature range where both the native and unfolded forms exist simultaneously. Researcher studies at UAMI have developed time-dependent kinetic equations to explain the unfolding and refolding mechanisms of TIM, G6PD, and laccase proteins. These equations include variables like as temperature and heating rates, which contribute to a more comprehensive comprehension of the hysteresis cycles seen in experiments. These discoveries provide valuable information on the thermodynamic and kinetic characteristics of these proteins, enhancing our understanding of protein folding and stability [7,25-30,32,33].
The main difficulties related to the irreversible thermal denaturation of proteins in aqueous solutions arise from the clumping together of unfolded polypeptide chains at elevated temperatures. Aggregation may result in irreversibility during the unfolding of proteins, making it challenging to use equilibrium thermodynamics to investigate protein denaturation. Irreversible unfolding curves are often found in oligomeric proteins that consist of subunits of moderate to large size.
Hysteresis cycles in protein unfolding-refolding profiles during temperature scanning are characterized by distinct, non-overlapping paths for unfolding and refolding [33].
This suggests that equilibrium conditions are not achieved inside the transition area. The difference between the temperature-dependent curves during heating and cooling results in a loop-shaped pattern, which is often associated with the sluggish kinetics of the unfolding and refolding operations. This occurs within a temperature range where both the native and unfolded states coexist in equilibrium. The protein's sluggish response speeds lead to distinct behaviors throughout the processes of heating (unfolding) and cooling (refolding), which ultimately result in hysteresis. Examining these cycles yields useful observations on the dynamics of protein unfolding and refolding processes, revealing the protein transitions.
Protein transition models: two-state mechanisms and their implications
Temperature-induced transitions in small proteins can often be represented, as a simpler two-state model involving only two molecular species: the native (N) and the unfolded (U) states. This model assumes that the populations of protein molecules are nearly at equilibrium at any temperature. Therefore, equilibrium thermodynamics can be used to determine the basic thermodynamic parameters of the unfolding process, such as the enthalpy and entropy changes associated with the transition from N to U. If differential scanning calorimetry data are available, it is also possible to calculate the heat capacity change, thus allowing for a complete description of the native−protein stability given by the Gibbs free energy change as a function of temperature.
A linkage between thermodynamics and kinetics can be easily found by writing the
unfolding and refolding reactions as first-order unimolecular reactions, since
the equilibrium constant for the N ↔ U transition is then expressed as
More complex models and irreversibility
In many cases, however, some complicating factors preclude a thermodynamic, straightforward analysis. For many large proteins (i.e., those with a molecular mass of about 25 kDa or larger), irreversible reactions, such as aggregation or chemical modification of some amino acid side chains, taking place when the protein is (partially) unfolded, prevent the recovery of the native structure and its biological activity. In some instances, aggregation can be avoided by decreasing protein concentration, thus making the system reversible; in other cases, even though the N to U process can be made to behave reversibly under low protein concentrations, the unfolding−refolding curves show that the overall system is far from equilibrium.
The first case reviewed below exemplifies how irreversibility has been dealt with in many early studies of protein thermal unfolding; in summary, data analysis has been carried out by assuming that irreversibility reactions occur more slowly than unfolding−refolding processes, and thus the equilibrium formalism can be applied to those data, with the inclusion of an irreversible step at the end of unfolding: N ↔ U → D, where D stands for an irreversibly denatured molecular form that can be the initiator of molecular aggregation.
Glucosamine-6-phosphate deaminase
The thermal unfolding of glucosamine-6-phosphate deaminase, an allosteric enzyme whose structure is described as a trimer of dimers (Mr = 178.2 kDa, Fig. 4), was studied by Hernández-Arana et al. using the scanning calorimeter at the Biophysical Chemistry Laboratory in UAMI. The unfolding/denaturation process of the enzyme was irreversible in the wide concentration range compatible with the experiments (0.6 to 7.3 mg/mL); however, analysis of scanning endotherms showed that the process is complex, involving several hexameric intermediates and, probably, two dissociated states. Evidence from this analysis suggested that some transitions are close to equilibrium, whereas at least one transition introduces irreversibility. Furthermore, applying an equilibrium formalism for data analysis seemed justified, provided scanning rates above 0.75 K/min were used.[7]
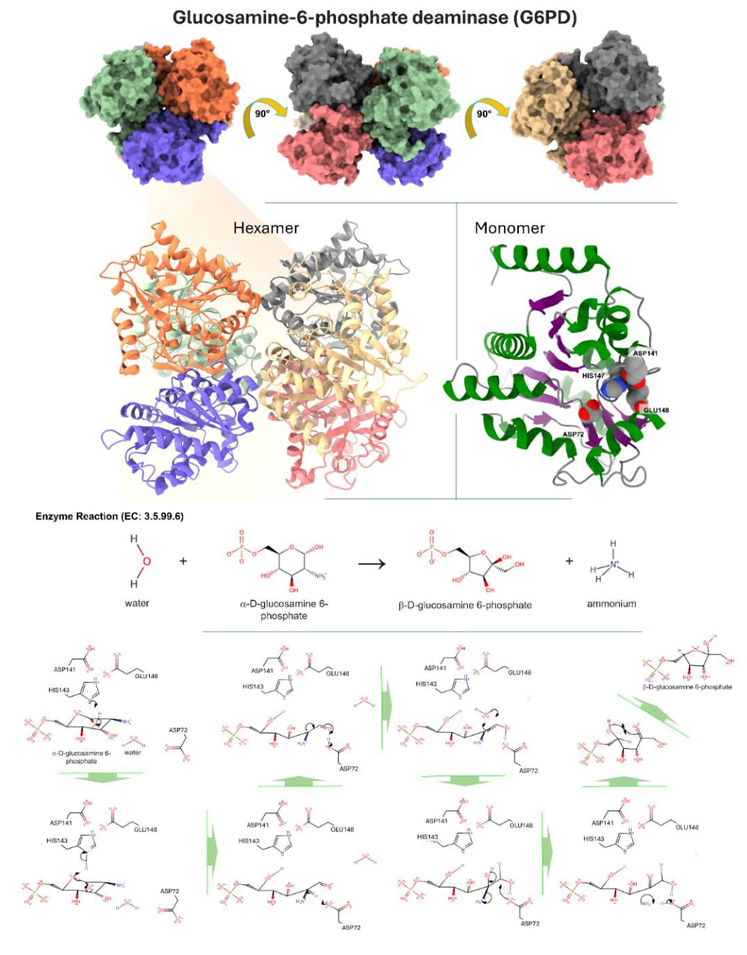
Fig. 4. Displays the three-dimensional structure of glucosamine-6-phosphate deaminase, which can be identified by its PDB code 1FS5 and UniProt code P0A759. The upper section depicts the hexameric arrangement of glucosamine-6-phosphate deaminase. The protein undergoes a 90° rotation in three different orientations, which emphasizes the presence of six monomers colored in bright green, blue, orange, red, gray, and warm yellow. The hexamer is presented as a cartoon image with the same color scheme, located in the bottom left corner. Glucosamine-6-phosphate deaminase is classified as belonging to the alpha/beta class. It has a 3-layer structure with an α/β/α sandwich architecture and a Rossmann fold topology (CATH: 3.40.50.1360). Furthermore, ithas a distinctive signal of glucosamine/galactosamine-6-phosphate isomerase (Prosite). The lower middle portion exhibits a monomer that emphasizes the amino acids in the active site: ASP72, ASP141, HIS147, and GLU148. The α-helices are shown in the color green, the β-sheets in the color purple, and the loops in the color light gray. The enzymatic reaction type and catalytic mechanism are given on the left. During the ring-opening process of α-D-glucosamine 6-phosphate, the hydroxyl group at C1 is deprotonated by HIS147. The oxyanion undergoes a collapse, resulting in the C-O link breaking. This process also leads to the deprotonation of HIS147 by the newly created oxyanion. ASP72 removes a proton from the carbon that is connected to the amino group. The carbanion that is formed initiates a rearrangement of the double bond, removing a proton from ASP72. Water undergoes electrophilic addition across the π link between carbon atoms C1 and C2. The water molecule approaches from the same side as ASP72. ASP72 works as a base by taking away a proton from the newly attached hydroxyl group. This gets rid of the ammonia in the substrate. When released, ammonia removes a proton from ASP72, forming ammonium and restoring the enzyme to its original state. The linear product spontaneously cyclizes outside the enzyme's active region, forming β-D-fructofuranose 6-phosphate and ammonium [34].
By assuming that six two-state sequential transitions are involved in the global process (two of which involve dissociation into subunits, eq. 1), the authors developed a set of equations describing the behavior of differential scanning calorimetry endotherms. Fitting to experimental data gave enthalpy changes and T m (i.e., the temperature at the maximum of the heat capacity curve) seemingly reasonable for the unfolding and dissociation values expected of protein subunits of similar size.
Triosephosphate isomerase
Triosephosphate isomerase from Saccharomyces cerevisiae (yTIM) is an enzyme that has a molecular weight of 54.52 kDa, with each subunit weighing 27 kDa (as shown in Fig. 5). Its biological function is to facilitate the reversible conversion between the triosephosphate isomers dihydroxyacetone phosphate and D-glyceraldehyde 3-phosphate. Hernández-Arana and colleagues have conducted research on this topic for more than twenty years, using circular dichroism as well as other calorimetric and spectrometric methods [25-28,32].
The triosephosphate isomerase dimer requires denaturation at around 63 °C, according to data reported by Hernández-Arana and colleagues. The unfolded state of yTIM is maintained when a temperature heating ramp of 0.2 °C/min is applied. Still, as seen in Fig. 6, a cooling ramp of 2 °C/min restores its original characteristics, indicating that there may be related events in the folding process. Some oligomeric proteins exhibit hysteresis cycles that may indicate a complex folding mechanism. This is backed up by research conducted between 2001 and 2022, which highlights the significance of dimer formation or stability in the folding and function of yTIM. Later on, similar findings were made in complex proteins, where functional areas impact the early folding process and impede folding. Even though these regions make folding more complicated, they may also affect other kinetic behaviors that are related to the system's stability.
The lack of hysteresis in lysozyme may be ascribed to its enduring monomeric structure and its two-state heat transition. This process exhibits a denaturation enthalpy (∆H) and heat capacity (∆Cp) that signify a substantial but reversible alteration in structure. Specifically, the thermodynamic parameters for lysozyme unfolding−refolding are within a range that leads to fast unfolding and refolding reactions within the transition region; therefore, the two-state system is very near to equilibrium at any temperature of the heating or cooling curves. In addition, this process reduces water-repellent surface contact and promotes effective and fast restoration of the original structure, preventing misfolding side reactions.
However, because of its dimeric structure and a more intricate denaturation process, yTIM displays noticeable hysteresis. The enthalpy change (∆H) required for denaturation of yTIM is much greater, indicating the need to disrupt a larger number of intermolecular connections throughout the process. In addition, the heat capacity (∆Cp) in yTIM is much higher, suggesting a notable change in the exposure of polar and nonpolar surfaces after denaturation. The higher ∆Cp value means that there are a lot of hydrophobic surfaces, which helps the unfolded subunits stick together. This makes it harder for them to get back to normal, which is what causes the hysteresis that we see.
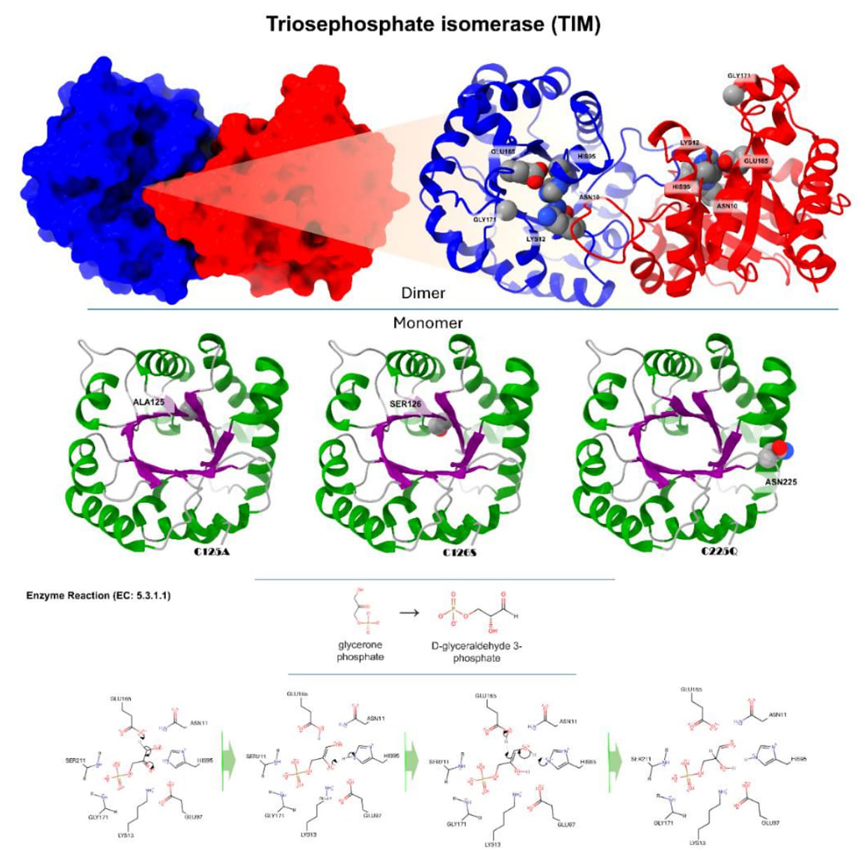
Fig. 5 Depicts the tridimensional configuration of triosephosphate isomerase (yTIM), which can be identified by its PDB code 1YPI and UniProt code P00942. The top part illustrates the dimeric configuration of yTIM. The monomers are shown in blue and red, while the dimer is illustrated in a cartoon depiction with the same color scheme. This highlights the active site amino acids: ASN10, LYS12, HIS95, GLU165, and GLY171. yTIM is categorized as an alpha/beta class protein, characterized by an Alpha/Beta Barrel structure, a TIM Barrel topology, and belonging to the Aldolase class I homology (CATH: 3.20.20.70). Moreover, it has a unique and recognizable molecular pattern and characteristics of the triosephosphate isomerase family (Prosite). The bottom portion displays a monomer with the "TIM barrel" fold. The secondary structural components are shown using α-helices in the color green, β-sheets in the color purple, and loops in the color light gray. This section focuses on three specific mutants: C125A, C126S, and C225Q. The enzymatic reaction type and catalytic mechanism are given on the left. By taking away a proton from the alpha-carbonyl carbon of glycerin phosphate, GLU165 acts as the catalytic base. HIS95 is deprotonated by the enolate. It is stronger for the hydrogen bond between the residue and the enol-transition state when HIS95 is deprotonated than when it is protonated. Now, HIS95 is the catalyst that helps the enol turn into an aldehyde while also selectively deprotonating C2. The formation of the final D-glyceraldehyde 3-phosphate is complete [34].
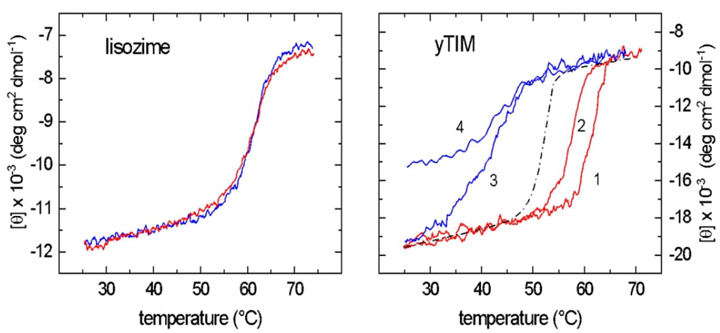
Fig. 6 Hysteresis landscape for yTIM unfolding investigations. (A) Diagram of the lysozyme's unfolding-refolding process, characterized by overlapping equilibrium curves. The graphs illustrate the fast unfolding−refolding kinetics that lead to a molecular system being very close to "equilibrium" at any temperature registered during the scanning. (B) Hysteresis graph associated with yTIM unfolding−refolding curves. In contrast to lysozyme illustrated in Figure A, the "equilibrium" unfolding and folding contours for yTIM are not equivalent. The 2.0 °C/min−heating and cooling cycle (curves 1 and 3) allows recovery of yTIM native structure and enzymatic activity. On the other hand, the heating and cooling process at 0.2 °C/min creates a misfolded state that makes it harder for yTIM to return to its original shape and structure, as shown in curves 2 and 4. For a two-state model, the apparent (hypothetical) equilibrium during the transition is denoted by the dashed line in the center. Figure redrawn from [25].
Hysteresis is a phenomenon in which systems delay their response to imposed stressors. This delay is caused by a bifurcation in the folding topography, as demonstrated by Hernández-Arana and colleagues' research on thermal unfolding and refolding in yTIM. In these experiments, a bistable system is shown, where the equilibrium state is defined by both the current and the beginning circumstances, acting as a memory for the system. Hysteresis occurs when the present and previous states of a system interact to affect its current state. Fig. 6 shows the results of the hysteresis-generating tests performed on yTIM, which include different ramps of heating and cooling. Curves 1 and 3 show quasi-perfect hysteresis behavior for yTIM, where the final state approaches the initial state but via a different pathway. On the other hand, curves 2 and 4 fall outside the hysteresis "zone," indicating non-ideal hysteresis. The dashed line shows the limit of ideal folded and refolded processes. The figure illustrates the intricate complexity of the folding domain. This finding provides further evidence that mechanisms other than folding might be influencing temperature-induced instability. When the functional dimer is formed, hysteresis takes place, as seen by the complex folding landscape of yTIM. Specifically, the stability of yTIM and the length of the catalytically active dimer half-life are determined by the temperature-dependent kinetic barrier to the unfolding mechanism. Significantly, hysteresis in TIM is reduced when heating and cooling rates are lower.
The hysteresis model of the TIM protein, as shown in Fig. 7, demonstrates a progressive temperature change at a rate of 2 °C per minute. This model exhibits unfolding at about 70 °C, followed by refolding by a different pathway. Approximately half of the protein remains in its folded configuration at a temperature of 45 °C, and its original shape is restored when the temperature is lowered to 25 °C. The enzyme activity recovers almost entirely, reaching roughly 95 % while maintaining the original structure [25].
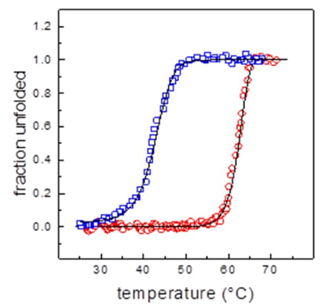
Fig. 7 Model for ideal yTIM hysteresis. Hysteresis is a characteristic that accounts for a system's delayed reaction to external forces. This phenomenon may occur when there is a split in the unfolded landscape, causing the system to have two stable conditions. This finding suggests that the system's equilibrium is influenced by both the initial and final states, demonstrating the system's ability to preserve structural information. The two graphs shown depict hysteresis curves, with red indicating unfolding and blue representing refolding, from experimental data; the separation between these two curves along the heating and cooling routes is noted, but it is important to highlight that the native state remains the same at the start and end of the cycle. Solid lines are simulations computed using a couple of differential equations (one, first-order reaction for unfolding and the other second-order for refolding), which together reproduce the characteristics of the hysteresis cycle, thus demonstrating that yTIM unfolding−refolding is a reversible far-from-equilibrium transition under kinetic control [33].
Recent studies on yTIM indicate that the hysteretic effect may be absent in some conditions. For example, mutations like ARG189-ASP225 or D225Q that disrupt the ionic interactions, or mutations like C126A and C126S1 that stop catalysis, could reduce this effect [26-28]. Hysteresis happens when folding transition states change because domain or monomer transitions affect complicated unfolding processes. Cohesive cooperation, on the other hand, makes refolding more efficient. Also, hysteresis can be seen in protein aggregation and association events, which shows how hard it is to get back to the original tertiary structure. This is particularly true for proteins like yTIM, which are very stable and have complex topologies like barrel superstructures. Hernández-Arana's current study on yTIM folding, supported by spectroscopic evidence, uses molecular dynamics simulations to clarify folding states. A first-order kinetics study showed that the structures of unfolded and refolded yTIM are different at different pH levels. This suggests that the unfolding and refolding processes are separate [32].
This is consistent with TIM's resistance to temperature and pH variations, indicating that it folds almost completely and returns to its original shape when refolded. The analysis showed a decrease in helix content and an increase in unordered structures over time, following first-order kinetics and consistent with changes in ellipticity values at fixed wavelengths. Rate constants derived from structural fractions matched those from circular dichroism data variations at specific wavelengths. Molecular dynamics simulations performed with AMBER and OPLS force fields captured essential elements of the unfolding process observed in circular dichroism experiments. The simulations demonstrated significant loss of helical regions and an increase in coil structures at high temperatures. Additionally, beta strands appeared more resilient at elevated temperatures. The unfolding process occurred faster at high pH, aligning with experimental observations. However, structural differences between low- and high-pH unfolded yTIM were relatively small in Molecular dynamics simulations, indicating the need for further refinement of Molecular dynamics simulations procedures to better match experimental data.
However, we believe that our progress in computational studies of protein unfolding can be beneficial in this field. Nonetheless, the task of estimating the fundamental process behind protein folding has not yet been tackled. In this context, we are working on a simple approach that allows us to accurately calculate the metastable states of protein unfolding in yTIM using Markov State Models (MSM) with PyEMMA [7,25-30,32,33]. This approach is based on computing the folded and unfolded states at different temperatures and pH using molecular dynamics simulations [35]. From this, the essential secondary structure conservation quantities are obtained and used to calculate the conformational stability of the protein. With this approach, we have successfully calculated the mechanisms of yTIM, representatives of the major structural classes, as well as small stability differences due to changes in solution conditions. This approach can permit us to understand the dynamic conformational behaviors before conducting in-vitro experiments.
Irreversible transition in proteins
Hernandez-Arana and colleagues' investigation of the laccase enzyme has provided valuable knowledge on its possible use in several industries and biotechnologies, including the food, textile, and environmental sectors. Laccase, seen in Fig. 8 and identified as PDB: 1GYC and UniProt: Q12718, can catalyze the oxidation of phenolic substances, making it very important in several industries [36].
Laccase is a particular type of enzyme that contains several copper atoms and is made up of 499 amino acids. Significant amounts of alanine (10.6 %) and glycine (7.8 %) make up this enzyme. It also has 4.0 % histidine residues (20 amino acids), with 2 % of them being located in the active region (Fig. 8). In addition, laccase has 12 % protonable residues, of which 4 % are exposed to the solvent on its surface. This exposure could have a significant impact on the enzyme's activity and folding state. For example, the laccase enzyme derived from Trametes versicolor has the highest level of activity at a pH of 3.5, whereas a pH of 6.5 is most favorable for maintaining its structural integrity. Ten of the twenty histidine residues are crucial because they coordinate the four copper ions inside the active site. These ten histidine residues are very important for keeping copper ions stable and allowing electron transfer processes for substrate oxidation to happen during the catalytic cycle. In addition, histidines play a vital role in maintaining the structural stability of the enzyme. They help to ensure that the enzyme is correctly folded and that the active site remains intact. Histidine residues connecting domains 1 and 3 operate as "structural staples," and the four coppers hold them together like glue (Fig. 8). The four coppers are made up of T1, T2, and T3 copper sites, which are linked by the conserved motifs HXHG, HXH, HXXHXH, and HCHXXXHXXXXM/L/F. Th
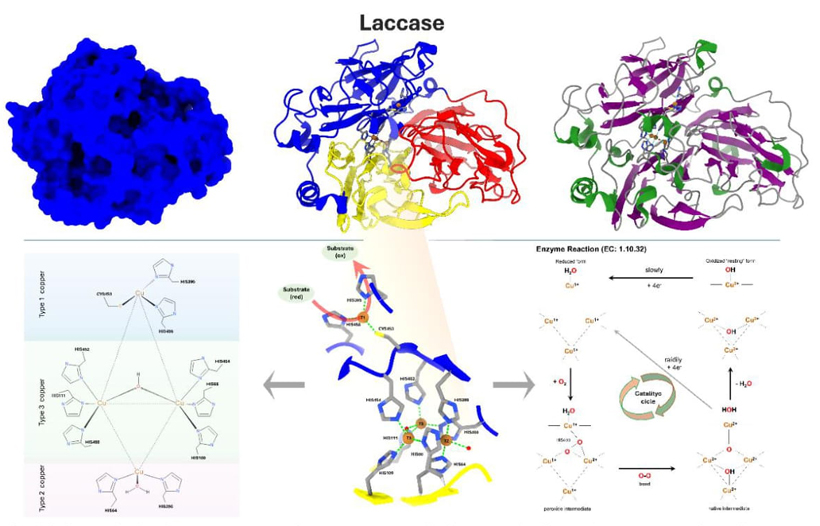
Fig. 8 This figure depicts the arrangement of Trametes versicolor laccase. The blue color represents the surface structure. The 3-domain structure is organized with Domain 1 embracing amino acids 1 to 130 and marked in yellow, Domain 2 embracing amino acids 131 to 300 and highlighted in red, and Domain 3 embracing amino acids 301 to 499 and highlighted in blue. The histidine and cysteine residues, coupled with the copper ions present in the active site, are highlighted. The laccase enzyme is seen on the far right, with its secondary structure color-coded: α-helices are represented in green, β-sheets in purple, and loops in light gray. Laccase is considered a member of the main β-class, with a β-Sandwich architecture, an immunoglobulin-like topology, and cupredoxin-blue copper protein homology, according to the CATH database (CATH: 2.60.40.420). In addition, it includes the Multicopper oxidase signatures (Prosite). The 2D representations in the lower left corner illustrate the catalytic site and its relationships. In the center of the 3D model, the catalytic location is displayed. This picture demonstrates the interconnection between CYS453, HIS452, and HIS454, which together constitute an electron route. Both pictures depict the OH molecule as an intermediate in the oxidation process, while the H2O molecule is the result of the redox reaction. The enzymatic reaction type and catalytic mechanism are given on the left. The first active site, called the "blue site," is close to the surface and is where organic substrates like phenols or aromatic and aliphatic amines go through oxidation. This lets the T1 copper ion get electrons through a bonded HIS458 residue. HIS395 and CYS453 complete the coordination of the T1 copper ion, which is not exposed to the solvent. The electrons that are taken out of the substrates are sent through two internal electron transfer pathways that start with CYS453, the T1 copper ligand. The electrons are then divided between HIS452 and HIS454, which respectively attach to the T3(a) and T3(b) copper ions in the second active site. In this process, oxygen molecules undergo reduction and join together to form water molecules. This site consists of a trinuclear copper cluster, which is composed of a T2 copper ion and two T3 copper ions organized in a triangle pattern. It is located between domains I and III and is attached to eight histidine residues and two water molecules. The T2 copper is tri-coordinated, forming bonds with HIS64, HIS398, and a single water molecule. The two T3 coppers exhibit tetra-coordination. T3(a) copper is coordinated by residues HIS111, HIS400, and HIS452, whereas T3(b) copper is coordinated by residues HIS66, HIS109, and HIS454. Furthermore, a water molecule is asymmetrically bound between the two T3 copper ions [34].
In 2012, Hernández-Arana and his colleagues conducted groundbreaking research using differential scanning calorimetry to assess the heat capacity of the enzyme laccase. Using this approach, they were able to investigate the thermal changes that occurred in the enzyme in both its native and unfolded forms under a variety of pH settings. The work of Hernández-Arana and others showed that the way copper ions are coordinated inside the laccase enzyme may affect how much heat it can hold (see Fig. 9). The data provided important information about the protein's thermal properties. For instance, laccase forms heat-denatured forms related to "molten-globule-like" structures, thus resembling other proteins such as α-lactalbumin, staphylococcal nuclease, and apomyoglobin. At low pH, the compact denatured state of laccase has residual structure and a low heat capacity, in contrast to the extended denatured state formed at pH 9.0.
The presence of copper ions and their interactions with the protein matrix can affect the magnitude in the protein's heat capacity, particularly during the thermal denaturation process. Therefore, heat capacity studies may provide insights into the thermodynamic characteristics of copper-containing proteins (see Fig. 9), especially when pH is imposed as an experimental variable.
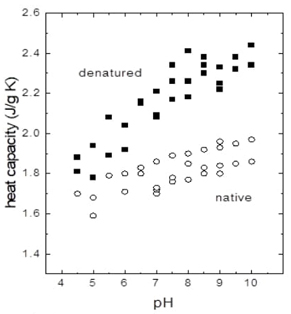
Fig. 9 The specific heat capacity (Cp) of a laccase protein in various structural states is plotted against the pH of the surroundings. Specific heat capacity is a characteristic that specifies how much thermal energy is needed to increase a substance's temperature by a certain amount. The graph shows four unique groupings of data points that reflect various laccase states: native-compact, native-extended, denatured-compact, and denatured-extended. The "native" state refers to the laccase in its folded and fully functioning form, usually at physiological pH values. As the pH varies, the protein's conformation may alter, making it more "compact" or "extended." With subsequent pH fluctuations, the laccase eventually achieves a "denatured" condition in which it loses its functional three-dimensional structure. The graph depicts how the specific heat capacity varies with conformational state and pH, which is critical for understanding laccase's stability and function under different situations. Redrawn from Biophysical Chemistry, 2012 [29].
The work by Hernández-Arana et al. showed that changes in the specific heat capacity of denatured laccase depend on pH. These changes show that the protonation or deprotonation of certain residues, like histidine residues, is important for keeping laccase's three-dimensional structure stable. The importance of pH in influencing the laccase enzyme's stability is highlighted. Histidine residues play an essential role in laccase folding since they are required for catalysis and tertiary structure. They operate as scaffolding, anchoring coppers at the active site and enhancing laccase's functional structure. They also serve as a route for the electron transport circuit used in laccase oxidation-reduction action. By looking more closely at specific heat capacity at different pH levels, Hernández-Arana and his colleagues learned more about the thermodynamic aspects of laccase folding and unfolding processes. This explains the irreversible processes involved in laccase folding, as seen in Fig. 9. When pH approaches 7.5, denatured laccase functions similarly to a completely solvated polypeptide chain, taking on a disordered structure comparable to an unfolded polypeptide chain. Denatured laccase retains some structural integrity between pHs 4.5 and 5.5. This demonstrates the significance of histidine residue protonation for enzyme stability. In the cited research, Equation 11 was used to explain how denatured laccase converts from compact to stretched configurations as a function of pH [29].
This equation shows the connection between changes in the logarithm of protonation equilibrium and the number of protons connected to the denatured laccase forms that are compact (C) and extended (E), which are shown as VE and VC, respectively. Its aim is to represent VE and VC as explicit pH functions., The constant Keq represents the equilibrium transition between VE and VC , allowing for a quantitative understanding of how specific amino acid residues affect the transition between these conformations. Furthermore, from analysis of experimental data, it was found that two histidine residues are most likely involved in the heat capacity change shown in Fig. 9.
In a 2016 paper published in Analytical Biochemistry, Hernández-Arana and his colleagues found that the excess molar heat capacity function (Cp) for the Myceliophthora thermophila laccase denaturation displays a complex profile that can be analyzed by four sequential irreversible steps (Fig. 10). These authors devised an analysis method consisting in step-by-step deconvolution of experimental C P curves that rendered kinetic constants and activation parameters for each of the four individual steps. Parameters derived from the analysis help us understand the thermal denaturation of the Myceliophthora thermophila laccase enzyme in more depth; moreover, these parameters can be used to extrapolate the denaturation kinetics constants to other temperatures, thus giving a way to estimate the stability of the enzyme under conditions in which it is employed in commercial processes [30].
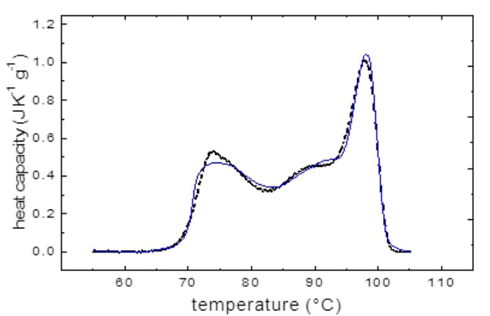
Fig. 10 Specific heat capacity (Cp) of laccase from Myceliophthora thermophila as a function of temperature. The graph shows a trend in which the specific heat capacity varies with temperature, with at least four peaks marking the temperature at which the laccase absorbs the greatest heat per unit rise in temperature. This suggests that four individual reaction steps take place during laccase denaturation. The graph's peaks show irreversible denaturation transitions, some of them involving structural transitions in which copper atoms participating in structural staples have probably been lost. The differential scanning calorimetry profile was recorded at a rate of 0.5 °C/min at pH 6.0; the solid line shows the experimental curve C p, obs curve (dotted line). The solid line, on the other hand, shows the calculated curve (C p, calc ), which was constructed by the deconvolution method described in reference 10.
Filamentous bacteriophage M13
In 2019, Hernández-Arana and colleagues conducted research that specifically examined the thermal cooperativity in the aggregation of the filamentous bacteriophage M13, commonly referred to as M13. Their study yielded valuable insights into the stability and dynamic characteristics of the M13 coat structure under varying environmental conditions [31].
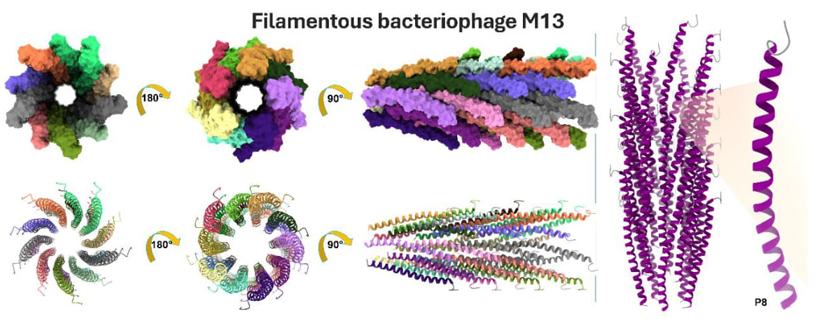
Fig. 11 Structural interpretation of the filamentous M13 phage capsid, which can be identified by its PDB code 2MJZ and UniProt code P69541. On the left, the multimeric organization of M13 is displayed, in which each individual subunit is determined by a P8 protein. The capsid undergoes a 90° and 180° rotation in three distinct orientations, revealing the existence of multimers that are colored in 35 different hues. The lower section displays the same orientations, but with ribbon representations in a cartoon-like manner. On the left, the M13 phage and the monomeric unit P8 are shown with their secondary structures color-coded. The α-helix is predominantly purple, while the N-ter and C-ter coil sections are depicted in gray. The M13 bacteriophage is classified as belonging to the Mainly Alpha class and has an Up-down Bundle architecture and single α-helix topology (CATH: 1.20.5.80), characterized by being engaged in coiled-coils or other heli x-helix interactions. Filamentous phages are elongated and semiflexible viruses composed of single-stranded DNA that have a unique affinity for infecting bacteria. The M13 phage, belonging to the Inoviridae family, has a length of roughly 1 micrometer and a diameter of around 7 nanometers. The P8 subunit consists of a single helix, six connections between helices, and beta turns. The structure is mostly formed of α-helical subunits that are stacked together in pentamers. These subunits exhibit a type II β-turn at the N-terminal. Each subsequent pentamer is separated by a vertical distance of 16.6-16.7 Å and an angular displacement of 36.1-36.6°. A repeated hydrophobic stacking pocket makes the subunit packing stronger. Each subunit adds a different hydrophobic residue to one of four pockets that are spread out along the subunit sequence.
The denaturation process of M13 exhibits notable changes in cooperativity in response to variations in pH and ethanol concentration. The denaturation process becomes more cooperative at elevated pH levels or greater ethanol concentrations. Specifically, the first denaturation phase gets more cooperative as the activation enthalpy for this step rises. Cooperativity is significantly enhanced by the presence of ethanol, particularly at values of 30 % and 50 %. The enhanced cooperativity is ascribed to the reinforcement of electrostatic contacts between DNA and proteins, which is caused by a reduction in the dielectric constant of the solvent [31].
P8 moieties have a vital function in modifying the arrangement of M13 bacteriophage molecules in response to surrounding conditions. Due to its versatility, the M13 coat structure has a high degree of malleability. When the virus is exposed to ethanol, it undergoes cooperative denaturation in more significant areas, demonstrating its capacity to adapt to environmental changes dynamically.
The M13 bacteriophage has remarkable heat stability compared to other viruses and nanostructures often used in research and applications. The M13 bacteriophage is characterized as a virus that can withstand high temperatures and the presence of high levels of ethanol without being affected. At higher temperatures, this virus undergoes denaturation, which occurs at a temperature higher than other filamentous and rod-like viruses, such as the rigid tobacco mosaic virus (TMV). The temperature at which denaturation is maximized (Tm) is twelve Celsius degrees higher than TMV at the same pH. Furthermore, the denaturation process of M13 exhibits a level of cooperativity equal to or greater than that of TMV under certain circumstances. The results indicate that the M13 bacteriophage has remarkable resistance to changes in temperature and shows a high degree of cooperation, making it a desirable option for a range of uses in biotechnology, materials science, and nanotechnology. The virus's ability to withstand extreme temperatures and environmental conditions creates opportunities for developing novel materials and technologies that may use its distinctive characteristics. The M13 bacteriophage has remarkable flexibility and adaptability to various pH levels and ethanol concentrations, enabling it to preserve its structural integrity and functionality even under diverse environmental circumstances. Due to its properties, it is well-suited for several applications in biotechnology and industry, including targeted medication delivery, gene therapy, and the formation of self-assembling nanostructures [31].
Conclusions
This work offers a comprehensive understanding of the dynamics and stability of important proteins by using modern biophysical methods such as differential scanning calorimetry, fluorescence, and circular dichroism. The research conducted at UAMI has provided valuable insights into the folding mechanics, stability, and possible biotechnological uses of proteins such as triosephosphate isomerase, Glucosamine-6-phosphate deaminase, laccase, and filamentous bacteriophage M13. Advancements like as deconvolution in differential scanning calorimetry analysis have facilitated a more thorough understanding of denaturation processes. The combination of multiples disciplines in this technique has significant promise for expanding the field of biotechnology and developing novel protein-based medicines.
A Tribute to Dr. Andrés Hernández Arana, a Trailblazer in the Physical Chemistry of Proteins in México
Dr. Andrés Hernández Arana is a well-regarded authority in the field of protein thermodynamics, with a remarkable career spanning five decades. He first studied biochemical engineering at IPN's National School of Biological Sciences. He then went on to get both a Master's and Doctorate in Chemistry from UAMI. Dr. Hernández Arana led the creation of the "Biofisicoquímica" (Biophysical Chemistry) research area in 1992 while serving as Head of the Chemistry Department at UAMI from 1989 to 1993. This area has evolved into a center for cutting-edge research, with a particular emphasis on investigating the stability of thermodynamic systems, studying the kinetics of proteins, and using sophisticated spectroscopic, calorimetric, and computational techniques. His scientific impact is demonstrated by the publication of more than 70 scientific papers and the guidance provided to 14 Master's and 13 Ph.D. students. The commitment of Dr. Hernández Arana has had a profound influence on Mexico's scientific community, creating an enduring heritage that continues to motivate successive generations of scholars.

