











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Transition metal-catalyzed transfer hydrogenation reactions are among the most effective methods for reducing unsaturated C=O, C=N, and C=C bonds. [1] This process offers significant advantages over traditional catalytic hydrogenation, as it eliminates the need for molecular hydrogen, high-pressure reactors, and harsh reaction conditions, thereby enhancing safety and convenience in both laboratory and industrial settings. In essence, transfer hydrogenation involves the transfer of a proton and a hydride from the donor molecule to the unsaturated substrate. Various hydrogen donors, such as alcohols,[2] formic acid,[3] and water,[4] have been employed in transfer hydrogenation reactions. These donors are not only inexpensive and readily available, but also much safer to handle than molecular hydrogen.
Among the non-H2 hydrogen sources in transfer hydrogenation, alcohols have emerged as effective hydrogen donors.[2] Isopropanol, in particular, is the most commonly used hydrogen source for reducing a wide range of substrates, as its oxidized product, acetone, can be easily removed from the reaction mixture. Unlike secondary alcohols, primary alcohols have an unfavorable redox potential and are generally oxidized to aldehydes, which are much more reactive than the acetone obtained from isopropanol.[5] Consequently, the use of primary alcohols as hydrogen sources is limited compared to secondary alcohols. However, primary alcohols such as methanol and ethanol are very attractive in terms of sustainability, as they can be obtained from renewable biomass, natural gas, or carbon dioxide.[6] The use of primary alcohols in transfer hydrogenation remains challenging due to various limitations. For instance, methanol produces formaldehyde as a by-product, which under strong reaction conditions can generate carbon monoxide, potentially poisoning the catalyst. Additionally, formic acid, another by-product, can neutralize the base commonly used in these catalytic transformations.[6]
In this context, over the last few decades, a wide variety of homogeneous transition metal catalysts, including noble metals such as ruthenium,[7-9] rhodium,[10-13] and iridium,[14-16] as well as more cost-effective options like nickel,[17] cobalt,[18] iron [19] and others, have been developed for catalytic transfer hydrogenation reactions.[19-21] In general, catalytic systems employing precious metals tend to perform effectively under relatively milder conditions compared to those based on non-precious metals. Interestingly, catalysts based on ruthenium have shown remarkable catalytic activity in the activation of alcohols and their subsequent use as a hydrogen source in the hydrogenation of carbonyl bonds.[7-9]
Since Grützmacher and co-workers reported the use of ethanol as the hydrogen source in transfer hydrogenation reactions in 2008,[22] numerous studies on ruthenium-catalyzed transfer hydrogenations utilizing ethanol have been conducted (Fig. 1). For example, in 2016, Khaskin et al. employed a Ru(II)-phosphine catalyst bearing a tridentate SNS ligand (A) for the transfer hydrogenation of esters using ethanol at 80 °C in the presence of a strong base (KOtBu) and toluene as the solvent.[23] Two years later, Weingart and Thiel reported on a Ru(II)-phosphine complex featuring a tridentate NNN ligand (B) for the reduction of aldehydes and ketones with ethanol using a strong base under a constant N2 flow at 40°C, yielding satisfactory results.[24] However, it's worth noting that the use of strong bases in transfer hydrogenation is not ideal due to the possibility of aldehydes undergoing ketone α-alkylation, leading to non-selective reactions.[25]
Subsequently, Bagh's group reported the use of a Ru(II) complex with a triazole-based ligand (C) as an effective catalyst for the transfer hydrogenation of a large number of aldehydes and ketones bearing various functional groups using both methanol and ethanol under aerobic conditions and relatively mild conditions in the presence of potassium carbonate, utilizing alcohol as both the solvent and the hydrogen source, resulting in excellent yields.[5] In 2022, Gong et al. developed a bidentate Ru(II)-NC (D) catalyst effectively catalyzing the transfer hydrogenation from azoarenes to hydrazoarenes with excellent selectivity.[25] They used 3 mol % of catalyst with a weak base and ethanol as the hydrogen source at 120 °C for 24 hours under an inert atmosphere.
Recently, Wang et al. also used the same Ru(II)-NC (D) catalyst for transfer hydrogenation of ketones using ethanol under weak base conditions. In this catalytic protocol, the reactions were carried out under an argon atmosphere at 120 °C for 5 hours using 10 mol% of catalyst. The results suggest that the reaction is almost as efficient when the reaction is carried out under air.[26] According to Albrecht et al., the transfer hydrogenation of ketones using ethanol could also be catalyzed by an air-stable, coordinatively unsaturated ruthenium (II) complex (E), reaching 66 % and 40 % yields with ethanol and methanol, respectively, after 24 hours at 80 °C.[27] Pratihar's research group has also reported the use of a waste shrimp shell-based tetrazene-Ru (II) p-cymene (F) catalyst for the transfer hydrogenation of aldehydes with ethanol.[28]
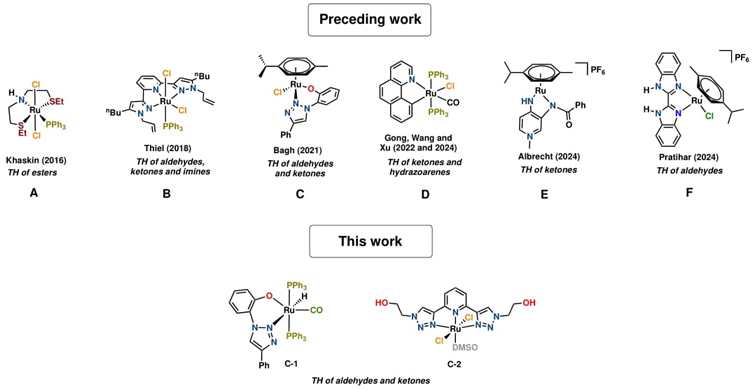
Fig. 1 Representative examples of well-defined ruthenium catalysts for the transfer hydrogenation (TH) of C=O, C=N and C=C bonds using ethanol as the hydrogen source.
As can be seen, the choice of ligands in ruthenium complexes as transfer hydrogenation catalysts plays a crucial role. In this regard, 1,2,3-triazoles, which can be easily synthesized via a copper(I)-catalyzed azide-alkyne cycloaddition reaction (CuAAC), have been extensively used as ligands in coordination chemistry. This is due to the presence of electron lone pairs on the triazole N2 and N3 atoms, which can coordinate with metal ions, forming stable metal complexes. Additionally, the predictable and modular composition of triazole-based ligands allows for fine-tuning of the steric and electronic properties of their metal derivatives by modifying the constituent substituent groups.
Motivated by the potential applications of ruthenium metal complexes and considering the importance of triazole ligands, we became interested in developing the chemistry of triazole-based ruthenium compounds. Ruthenium and triazole moieties form an effective combination for producing bioorganometallic compounds with potential biological and catalytic properties. In addition, given the growing demand for environmentally friendly methodologies and inspired by recent advancements in catalytic processes utilizing ethanol as a hydrogen source in transition metal-catalyzed transfer hydrogenations, herein, we disclose the synthesis and full characterization of two new air-stable triazole-based Ru(II) complexes and their catalytic evaluation in the homogeneous transfer hydrogenation of ketones and aldehydes under relatively mild conditions (1 mol% [Ru], 10 mol % of K2CO3 90 °C, 3 and 24 h, in air), using ethanol as both the hydrogen source and solvent. This process produces secondary and primary alcohols in good to excellent yields with high selectivity.
Experimental
General considerations
Unless otherwise noted, all experiments were performed in air. All solvents were purchased from commercial suppliers and used without further purification. All other chemicals and filter aids were reagent grade and were used as received. Column chromatography was performed on silica gel (Merck, 230-700 mesh). Elemental analyses were performed in a Thermo Scientific Flash 2000 elemental analyzer. NMR experiments were recorded at 300 K on Bruker Avance DMX-500 (500 MHz) spectrometer using TMS or residual proton solvents as internal standard; and H3PO4 as external standard. The deuterated solvent used was CDCl3 and DMSO-d6; chemical shifts (δ) are quoted in ppm and coupling constants in Hz; to indicate the multiplicity of the signals of 1H NMR spectra, the following abbreviations have been used: (s) singlet, (d) doublet, (t) triplet, (at) apparent triplet, (m) multiplet, (dd) double doublet, (bs) broad signal. Catalysis products were quantified with a GC-MS Agilent 6890N chromatograph equipped with a 30 m DB-1MS Agilent capillary column, coupled to an Agilent Technologies 5973 Mass Spectrometer equipped with an Inert Mass Selective Detector. FTIR spectra of the samples were recorded using a Perkin-Elmer 600 spectrometer using the attenuated total reflectance (ATR) method. The absorbance peaks are reported in reciprocal centimeters (cm-1). Mass Spectrometer equipped with an Inert Mass Selective Detector. Mass spectra were recorded on mass spectrometer model micrOTOF II (Bruker Daltonics Inc.) using the Compass platform (otofControl and DataAnalysis from Bruker Daltonics Inc.). Spectra were acquired in positive mode with a capillary voltage of 4500 V, nebulizer gas: 0.5 Bar, drying gas 4.0 L/min and a drying temperature of 150 °C.
Synthesis of ligands L1 and L2
Synthesis of ligand L1 (2-(4-phenyl-1H-1,2,3-triazol-1-yl)phenol)
Ligand L1 was synthesized according to the literature procedure.[5,29] Sodium ascorbate (0.200 g, 1.0 mmol) and CuSO4·5H2O (0.025 g, 0.1 mmol) were added to a solution of 2-azidophenol (1.340 g, 10.0 mmol) in a 1:1 mixture of water and tert-butanol (50 mL). Thereafter, phenylacetylene (1.32 mL, 11.0 mmol) was added dropwise. The reaction mixture was stirred at 90 °C for 24 h. The resultant mixture was then added to ice-cold water (100 mL), resulting in the formation of a yellow-green precipitate. The yellow-green solid was isolated by filtration and then purified by column chromatography using silica gel and a 1:1 mixture of ethyl acetate and hexanes as eluent. The final product was isolated in 82 % yield as a light-yellow pure solid. 1H NMR (500 MHz, CDCl3) δ 9.89 (s, 1H), 8.30 (s, 1H), 7.93 - 7.89 (m, 2 H), 7.52 - 7.45 (m, 3H), 7.44 - 7.37 (m, 1H), 7.32 (ddd, J = 8.3, 7.3, 1.5 Hz, 1H), 7.22 (dd, J = 8.3, 1.3 Hz, 1H), 7.03 (ddd, J = 8.1, 7.3, 1.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 149.6, 147.9, 129.9, 129.7, 129.2, 129.1, 126.2, 122.9, 120.4, 119.7, 119.5. ESI-TOF: 238.0971 [(M+H)]+ (100 %), calculated for C14H11N3O: 237.0902.
Synthesis of ligand L2: (2-(4-(6-(1-(2-hydroxyethyl)-1(4,2,3(2-triazol-4-yl)pyridin-2-yl)-1H-1,2,3-triazol-1-yl)ethan-1-ol)
Ligand L2 was synthesized in three steps using an adapted procedure documented previously.[30]
Synthesis of 2,6-bis((trimethylsilyl)ethynyl)pyridine: In the first step, 2,6-dibromopyridine A (2.13 g, 8.95 mmol), CuI (200 g, 1.05 mmol), and Pd(PPh3)4 (620 g, 0.88 mmol) were added to a 100 mL Schlenk flask. Then, 36 mL of anhydrous THF and 6 mL of Et3N were added. The resulting yellow solution was stirred at room temperature under a nitrogen atmosphere for 15 minutes. Next, ethynyltrimethylsilane (2.19 g, 22.9 mmol) was added dropwise via syringe into the solution, and the reaction mixture was stirred under a nitrogen atmosphere for 10 hours. After the reaction time, the resulting mixture was added to a separatory funnel along with 10 mL of 1 M NH4Cl aqueous solution and extracted with dichloromethane (3 x 20 mL). The organic phase was dried with Na2SO4, filtered, and evaporated under reduced pressure, generating a brown solid. This solid was purified by flash chromatography using silica gel and a 5:95 mixture of EtOAc/Hexanes as eluent. The product, 2,6-bis((trimethylsilyl)ethynyl)pyridine, was isolated in 98.1 % yield as a light-brown solid. 1H NMR (500 MHz, CDCl3): δ 7.57 (dd, J = 8.1, 7.5 Hz, 1H), 7.36 (dd, J = 7.8, 0.3 Hz, 2H), 0.24 (s, 16H). 13C NMR (126 MHz, CDCl3) δ 143.5, 136.3, 126.8, 103.3, 95.5, 77.4, 77.2, 76.9, -0.2.
Synthesis of 2,6-diethynylpyridine, B: In the second step, 2,6-bis((trimethylsilyl)ethynyl)pyridine (1 g, 3.31 mmol) and 15 mL of dichloromethane were added to a round-bottom flask equipped with a magnetic stirrer. NaOH (530.3 mg, 13.25 mmol) dissolved in 15 mL of methanol was then added to the flask. The reaction mixture was stirred at room temperature for 30 minutes. After this time, the solvent was removed under reduced pressure, and the crude product was immediately purified by chromatography using silica gel and a 10:90 mixture of EtOAc/Hexanes as eluent. The product 2,6-diethynylpyridine, B, was isolated in 98 % yield as a light-yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.62 (t, J = 7.8 Hz, 1H), 7.42 (d, J = 7.8 Hz, 2H), 3.14 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 142.8, 136.6, 127.2, 82.2, 77.8.
In the final step, a 100 mL round-bottom flask was charged with 2,6-diethynylpyridine (250 mg, 1.966 mmol), CuSO4·5H2O (99.68 mg, 0.399 mmol), sodium ascorbate (158.18 mg, 0.7984 mmol), and 7.5 mL of tBuOH. Then, 2-azidoethan-1-ol (3.93.7 mg, 4.52 mmol) in 7.5 mL of water was added. The reaction mixture was heated at 100 °C for 24 hours. After the reaction time, the resulting mixture was added to a separatory funnel along with 10 mL of 1 M NH4Cl/EDTA aqueous solution and extracted with ethyl acetate (3 × 20 mL). The organic phase was dried with Na2SO4, filtered, and evaporated to approximately 2/3 of the total volume under reduced pressure. A precipitate formed, which was filtered and washed with a minimum volume of cold ethyl acetate. Ligand L2 was isolated in 86 % yield as a white crystalline solid. 1H NMR (500 MHz, DMSO) δ 8.62 (s, 2H), 7.97 (s, 3H), 5.10 (s, 2H), 4.51 (t, J = 5.4 Hz, 4H), 3.86 (s, 4H), 3.33 (s, 1H). 13C NMR (126 MHz, DMSO) δ 150.0, 146.9, 138.1, 123.8, 118.2, 59.8, 52.5. ESI-TOF: 302.1359 [(M+H)]+ (100 %), calculated for C13H15N7O2: 301.1287.
Synthesis of ruthenium complexes C-1 and C-2
Synthesis of ruthenium complex C-1
A 100 mL round-bottom flask equipped with a magnetic stirring bar was charged with 200.0 mg (0.218 mmol) of [Ru(PPh3)3H2CO]31, 39 mg (0.218 mmol) of ligand L1 and 20 mL of THF. To this solution, 46 μL (0.744 mmol) of Et3N was added, and the resulting reaction mixture was heated at 67 °C for 48 hours. The resulting solution was evaporated under reduced pressure, and the residue was purified by flash chromatography on silica gel using EtOAc/hexanes (20:80) as the eluent. Complex C-1 was isolated as a white-off powder in 68 % yield. 1H NMR (500 MHz, CDCl3) δ 7.65 - 7.58 (m, 14H), 7.45 - 7.41 (m, 2H), 7.39 - 7.33 (m, 1H), 7.31 (s, 1H), 7.25 - 7.14 (m, 18H), 6.65 (ddd, J = 8.5, 6.9, 1.8 Hz, 1H), 6.50 (dd, J = 8.4, 1.4 Hz, 1H), 6.09 (dd, J = 8.0, 1.8 Hz, 1H), 6.03 (ddd, J = 8.1, 6.9, 1.4 Hz, 1H), -11.40 (t, J = 19.9 Hz, 1H).13C NMR (126 MHz, CDCl3) δ 204.7, (t, J= 15 Hz), 160.7, 147.3, 134.8 (at, J = 20.4 Hz), 134.2 (at, J = 6.0 Hz), 130.4, 129.1, 128.8, 128.3 (d, J = 5.4 Hz), 127.8 (at, J = 4.6 Hz), 126.2, 125.9, 125.8, 120.4, 116.9, 112.5. 31P{1H} NMR (126 MHz, CDCl3) δ 40.9. ESI-TOF: 892.1745 [(M+H)]+ (100 %), calculated for C51H41N3O2P2Ru: 891.1742. Anal. Calcd. for C-1, C51H41N3O2P2Ru: C, 68.76; H, 4.64; N, 4.72. Found: C, 68.75; H, 4.78; N, 4.75.
Synthesis of ruthenium complex C-2
A 100 mL round-bottom flask equipped with a magnetic stirring bar was charged with 117.4 mg (0.24 mmol) of [RuCl2(CH3SOCH3)4], 100 mg (0.24 mmol) of ligand L2, and 10 mL of THF. The resulting reaction mixture was heated at 67 °C for 24 hours. The solution was then evaporated under reduced pressure, and the residue was purified by flash chromatography on silica gel using CH3OH/acetone (50:50) as the eluent. Complex C-2 was isolated as an orange solid in 81.9 % yield. 1H NMR (500 MHz, DMSO) δ 9.15 (s, 1H), 8.19 (d, J = 8.3 Hz, 1H), 8.12 (t,1H), 4.59 (t, J = 5.1 Hz, 2H), 3.89 - 3.83 (m, 2H), 3.53 (s, 3H). 13C NMR (126 MHz, DMSO) δ 146.30, 137.20, 125.67, 118.24, 59.38, 54.07, 46.63, 23.28. Anal. Calcd. for C-2, C15H21Cl2N7O3RuS: C, 32.67; H, 3.84; Cl, 12.86; N, 17.78. Found: C, 32.71.; H, 4.01; N, 17.59. The ESI-TOF mass spectra of C-2 exhibited an [(M-Cl) + K]+ ion peak at m/z 556.05 calculated for C15H21ClN7O3RuS.
Catalytic experiments
The reactions were performed in 25 mL reaction tubes equipped with an inner magnetic stirring bar using a Carousel 12 Plus reaction station. The tubes were charged with a mixture of the corresponding Ru catalyst (0.01 mmol), ketone or benzaldehyde derivatives (1 mmol), base (0.1 mmol), and 4 mL of ethanol or isopropanol. The reaction mixture was stirred and heated at 90 °C or at room temperature for different reaction times. After the prescribed reaction times, the resulting mixtures were cooled to room temperature, filtered through Celite, and analyzed by GC-MS. After taking an aliquot for GC-MS analysis, selected crude products were purified by silica gel column chromatography using ethyl acetate/hexanes as the eluent and analyzed by 1H NMR.
Results and discussion
Synthesis and characterization of ruthenium complexes C-1 and C-2
Bidentate ligand L1 was synthesized using an adapted procedure documented previously,[5] utilizing commercially available 2-aminophenol and ethynylbenzene, as outlined in Scheme 1. L1 was obtained in two steps through Sandmeyer/click copper-catalyzed azide−alkyne reactions and isolated as a light-yellow solid with a yield of 82 % after purification by flash chromatography. This ligand was further characterized by 1H and 13C{1H} NMR spectroscopy and structurally confirmed by direct comparison of previously reported data (full details are provided in the experimental section).
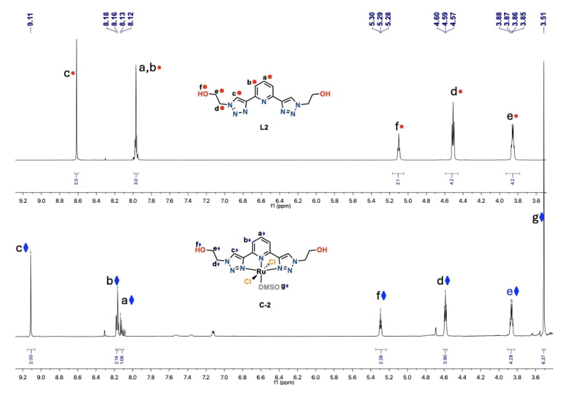
Ligand L2 was easily obtained owing to the flexibility of copper-catalyzed azide−alkyne cycloaddition method. Consequently, the reaction between 2-azidoethanol and 2,6-diethynylpyridine yielded the desired tridentate ligand L2 as a white solid in a good yield of 86 % after purification by chromatographic column, as illustrated in Scheme 2. Due to the C 2 -symmetry in L2, only a few resonances were observed in the 1H NMR spectrum (SI, Fig. SI12). The spectrum of L2 displayed a characteristic singlet at 8.62 ppm assigned to the proton of the triazole moiety, as well as a multiplet at 7.99 - 7.9 ppm resulting from the overlap of the pyridyl signals. The methylene bonded to the -OH group appeared as an apparent quadruplet at 3.86 ppm, and the methylene attached to the triazole ring was observed as a triplet at 4.51 ppm, while the alcohol moiety presented a triplet signal at 5.10 ppm. Additionally, 13C{1H} NMR study and mass spectrum of L2 confirmed the formation of the tridentate L2 (see SI, Fig. SI13).
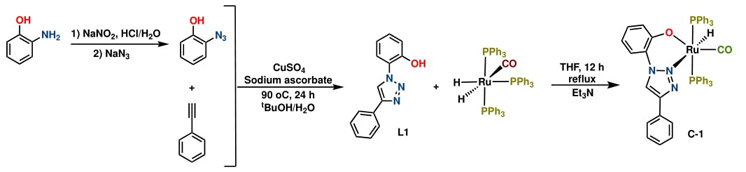
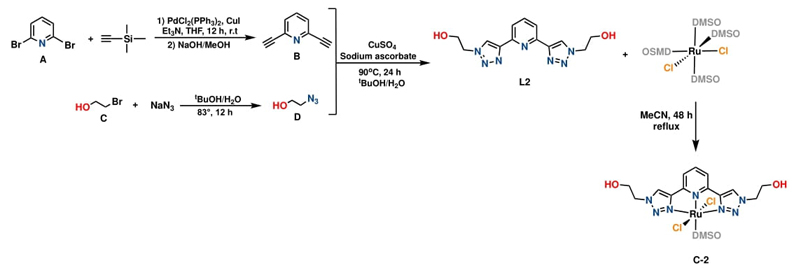
The ruthenium (II) complex C-1 was obtained through the facile coordination of L1 with [RuH2(PPh3)3CO] in the presence of Et3N, resulting in an air-stable off-white solid with a yield of 68% (Scheme 1). Similarly, the reaction of the tridentate N-donor ligand L2 with [RuCl2(DMSO)4] in tetrahydrofuran at reflux temperature yielded air-stable complex C-2 as an orange solid in 81.9% yield (Scheme 2).
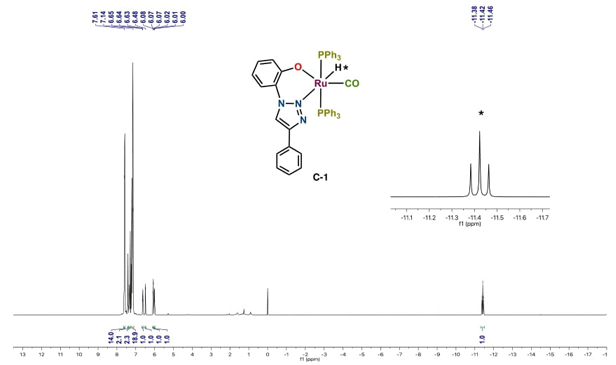
The identities of the newly synthesized ruthenium (II) complexes C-1 and C-2 have been confirmed through multinuclear NMR (1H, 13C and 31P for C-1 only), ESI-TOF mass spectrometry, and satisfactory elemental analysis. As expected, the 1H and 13C NMR spectra of bidentate ruthenium (II) complex C-1 are consistent with C 1 -symmetry. In the 1H NMR spectrum of C-1, a triplet at -11.42 ppm with 2JH-P = 19.9 Hz is observed, attributed to the Ru-H signal, consistent with its cis-position relative to two equivalent PPh3 groups (Fig. 2). Furthermore, signals for aromatic protons were observed in the range of 6-7.66 ppm, integrating to 40 protons. The proposed structure of C-1 was also further confirmed by 13C{1H} NMR, which exhibited a triplet signal for the CO ligand due to its coupling with two equivalents, mutually cis-phosphorus nuclei at 204.5 ppm (2JC-P= 15 Hz). Additionally, the phosphorus atoms of the PPh3 groups appeared as a singlet at 40.9 ppm in the 31P{1H} NMR spectrum, confirming their equivalence due to their mutual transposition. The resulting mass spectrum is consistent with the formation of complex C-1, showing the molecular ion ESI-TOF: 892.1745 [(M+H)]+ (100%), calculated for C51H41N3O2P2Ru: 891.1742. The CO stretching frequency for C-1 was observed at 1922.1 cm-1 and it is indicative of strong ( Ru-CO backbonding. Results from the elemental analysis of C-1 also validate the proposed structural formulation.
The Ru(II) tridentate pincer complex (C-2) was characterized in solution by 1H and 13C NMR, with its proton and carbon resonances shifting to higher frequencies compared to those of the free ligand L2 (vide supra) due to coordination to the metal center. Additionally, a new signal appears at 3.5 ppm as a singlet in the 1H NMR spectrum, integrating to 6 protons, which is assigned to a coordinated molecule of dimethyl sulfoxide to the ruthenium metal center, thus confirming the formation of the desired C 2 -symmetric complex C-2. In the 13C NMR, the signal of the carbon from coordinated dimethyl sulfoxide is observed at 46.57 ppm. In Fig. 3, a comparison of the 1H NMR spectra of the free ligand L2 and C-2 is presented. The ESI-TOF mass spectra of C-2 exhibited an [(M-Cl) + K]+ ion peak at m/z 556.05 calculated for C15H21ClN7O3RuS, and the elemental analysis also confirmed its chemical composition. Principio del formulario.
Catalytic activity
With both ruthenium pincer complexes in hand, their catalytic activity in the transfer hydrogenation of ketones and aldehydes using primary (EtOH) and secondary (isopropanol, IPA) alcohols as hydrogen donors was then examined and compared. We began the investigation by evaluating the activity of both ruthenium complexes (C-1 and C-2) in the catalytic transfer hydrogenation reaction of 4-bromoacetophenone, using K2CO3 as a base in either ethanol or isopropanol (Table 1). Among the screened complexes, C-2 was the most efficient catalyst in both alcohols, affording the 1-(4-bromophenyl)ethan-1-ol product in 89.3 % and 76.2 % yields in ethanol and isopropanol, respectively, at 90 °C for 24 h (entries 2 and 4). The distinctive reactivity observed in the C-1 and C-2 complexes bearing bidentate and tridentate ligands was attributed to the electronic effects and the thermal stability provided by the mer-tridentate pincer ligand to the catalytic compound C-2.
On the other hand, comparing the performance of C-2 in both alcohols, it was better in ethanol, despite isopropanol being a better hydrogen transfer agent than ethanol (see Table 1, entries 2 and 4). This was attributed to the low solubility of complex C-2 in the secondary alcohol. Based on this result, we decided to conduct a reaction of 4-bromoacetophenone in the presence of K2CO3 using a mixture of IPA:H2O (7:3), considering that C-2 is highly soluble in water. However, the yield remained poor (58 %) (entry 5). Reactions carried out either at room temperature or without a catalyst did not show any conversion (entries 7 and 8). Additionally, GC-MS monitoring of the reactions in ethanol revealed the formation of acetaldehyde, resulting from the dehydrogenation of ethanol, at the beginning of the reaction during the induction period (6-12 h). Subsequently, ethyl acetate was produced. We propose that the induction period is related to the generation of hydrides necessary for catalysis, and then acetaldehyde reacts with ethanol to produce ethyl acetate and release hydrogen.
Table 1 Transfer hydrogenation reaction of 4-bromoacetophenone and either ethanol or isopropanol catalyzed by ruthenium complexes C-1 and C-2 a.
|
| |||||
| Entry | [Ru] | Hydrogen source | T (oC) | T (h) | Yield of 2 (%)b |
| 1 | C-1 | IPA | 90 | 24 | 70.5 |
| 2 | C-2 | IPA | 90 | 24 | 76.2 |
| 3 | C-1 | Ethanol | 90 | 24 | 33.5 |
| 4 | C-2 | Ethanol | 90 | 24 | 89.3 |
| 5 | C-2 | IPA/H2O, 7:3 | 90 | 24 | 58 |
| 6 | C-2 | ethanol | 90 | 12 | 51.3 |
| 7 | C-2 | ethanol | r.t. | 24 | ND |
| 8 | None | ethanol | 90 | 24 | ND |
aReaction conditions: 4-bromoacetophenone (1.0 mmol), [C-2] (1 mol%), EtOH (4.0 mL), K2CO3 (0.1 mmol). bYields were determined by CG-MS and are the average of two independent runs. ND = Not detected. r.t.= room temperature.
As is well known, transfer hydrogenation reactions are strongly dependent on the base used. Thus, various bases were tested under the optimized reaction conditions using the best-found catalyst, C-2 (Table 2). The results obtained revealed that Cs2CO3 and K2CO3 afford the best yields (Table 2, entries 1 and 2). Since K2CO3 is cheaper than Cs2CO3, the former will be used in the following experiments.
Table 2 Transfer hydrogenation reaction of 4-bromoacetophenone with EtOH catalyzed by ruthenium complex C-2 using different bases.a
|
| ||
| Entry | Base | Yield of 2 (%)b |
| 1 | K2CO3 | 86.3 |
| 2 | Cs2CO3 | 86.6 |
| 3 | Na2CO3 | 78.1 |
| 4 | Li2CO3 | 85.1 |
| 5 | NaOH | 50.0 |
| 6 | Et3N | 18.5 |
aReaction conditions: 4-bromoacetophenone (1.0 mmol), [C-2] (1 mol%), EtOH (4.0 mL), base (0.1 mmol) for 24 h. bYields were determined by CG-MS and are the average of two independent runs.
To explore the effectiveness of complex C-2, the transfer hydrogenation reaction was also performed under the above reaction conditions with different catalyst loadings (Table 3). When 1 mol % of C-2 was used, 1-(4-bromophenyl)ethan-1-ol was obtained in 86.3 % yield after 24 hours (entry 3). Reducing the amount of catalyst to 0.1 and 0.5 mol % (entries 1 and 2) resulted in low to moderate yields of the alcohol product in 24 hours. Consequently, a catalyst loading of 1 mol % was used for further studies.
Table 3 Effect of catalyst C-2 loading.a
aReaction conditions: 4-bromoacetophenone (1.0 mmol), [C-2] (0.1-1 mol%), EtOH (4.0 mL), K2CO3 (0.1 mmol) for 24 h. bYields were determined by CG-MS and are the average of two independent runs.
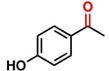
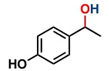
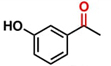
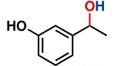
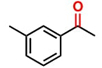
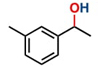
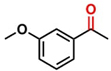
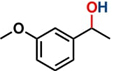
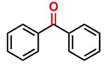
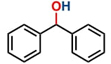
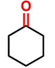
Hence, considering the optimized reaction conditions (1 mol % of C-2, 10 mol % K2CO3, at 90 °C for 24 hours in a closed vial and encouraged by these results, we turned our attention to extend the scope of this reaction to a series of substituted acetophenones to examine their electronic and steric effects (Table 4). Interestingly, variation of the electronic nature of the aromatic ring had little impact on the reaction efficiency and as a result, neutral (entry 1) along with electron-withdrawing and electron-donating substituents at para-positions provided the desired alcohol products in good yields (entries 2-7). On the other hand, meta-substituted acetophenones (entries 8-10) were hydrogenated slightly less effectively than the corresponding para-substituted derivatives (entries 5-7). Likewise, a good yield was achieved for the transfer hydrogenation of diaryl ketones. For instance, benzophenone yielded the desired reduced product, diphenylmethanol, with an 85 % yield (entry 11). Similarly, the use of aliphatic ketones, such as cyclohexanone, gave the corresponding secondary alcohol in a good yield (entry 12).
Table 4 Transfer hydrogenation reaction of ketones with EtOH catalyzed by ruthenium complex C-2.a
|
| ||||
| Entry | Ketone (1) | Alcohol (2) | Yield (%) | TOF (h-1) |
| 1 |
|
|
88.5b | 3.68 |
| 2 |
|
|
89.3b/84c | 3.72 |
| 3 |
|
|
89.0b/83.2c | 3.70 |
| 4 |
|
|
90.1b/85c | 3.75 |
| 5 |
|
|
93b/89.0c | 3.75 |
| 6 |
|
|
88.7b/86c | 3.69 |
| 7 |
|
|
87.9b | 3.66 |
| 8 |
|
|
78.9b/76.2c | 3.28 |
| 9 |
|
|
84.8b | 3.53 |
| 10 |
|
|
85.8b/81c | 3.57 |
| 11 |
|
|
85b | 3.54 |
| 12 |
|
|
79.9b | 3.32 |
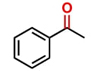
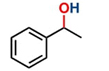
aReaction conditions: Acetophenone derivative (1.0 mmol), [C-2] (1 mol%), EtOH (4.0 mL), K2CO3 (0.1 mmol) for 24 h. bYields were determined by CG-MS and are the average of two independent runs. cYields of isolated compounds.
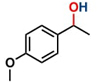
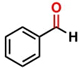
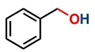
Thereafter, the transfer hydrogenation of aldehydes with ethanol was investigated under the optimized reaction conditions found for ketone reductions (Table 5). First, benzaldehyde was tested, and complete conversion with a quantitative yield of benzyl alcohol was obtained. Based on this result, we decided to optimize the reaction time and performed the hydrogenation of benzaldehyde at different times: 24, 12, 6, and 3 hours. Excellent yields were achieved at 24 hours (100 %, Table 5, entry 1) ,12 hours (100 %, Table 5, entry 2) and 6 hours (100 %, Table 5, entry 3), while at 3 hours the yield of the product was very poor (31 %, Table 5, entry 4). As expected, the reduction of aldehydes was easier than that of ketones, completing the reaction in only 6 hours. Then, we examined some substituted benzaldehydes using 1 mol % of C-2 and 10 mol % of K2CO3 at 90 °C for 6 hours, generating the primary alcohol with excellent yields.
Table 5 Transfer hydrogenation reaction of aldehydes with EtOH catalyzed by ruthenium complex C-2.a
|
| ||||
| Entry | Ketone (1) | Alcohol (2) | Yield (%) | TOF (h-1) |
| 1b* |
|
|
100c | 4.16 |
| 2b** |
|
|
100c | 8.33 |
| 3b*** |
|
|
100c | 16.6 |
| 4b**** |
|
|
31c | 10.3 |
| 5 |
|
|
100c | 16.6 |
| 6 |
|
|
95c/91d | 15.8 |
| 7 |
|
|
97.5c/92d | 16.25 |
| 8 |
|
|
99c | 16.5 |
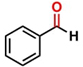
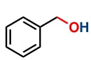
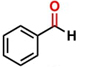
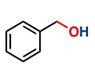
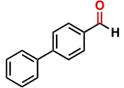
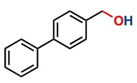
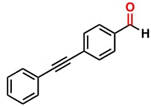
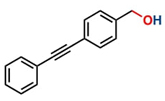
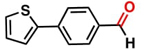
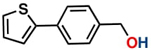
aReaction conditions: Benzaldehyde derivative (1.0 mmol), [C-2] (0.1-1 mol%), EtOH (4.0 mL), K2CO3 (0.1 mmol) for 6 h. bReaction conditions: Benzaldehyde (1.0 mmol), [C-2] (1 mol%), EtOH (4.0 mL), K2C03 (0.1 mmol) for: *24 h, ** 12h, ***6 h and ****3 h. cYields were determined by CG-MS and are the average of two independent runs. dYields of isolated compounds.
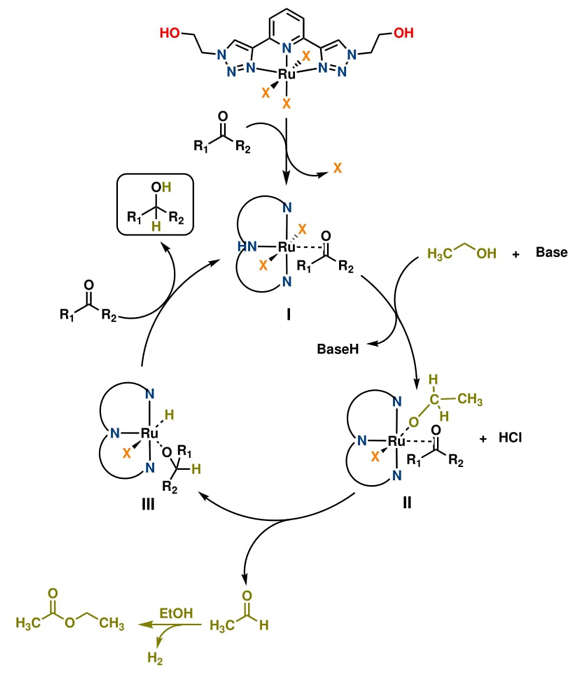
Based on literature reports,[32,33] a comprehensive mechanistic proposal for the hydrogenation of carbonyl bonds catalyzed by the ruthenium complex (C-2) is illustrated in Scheme 3. The mechanism likely involves the release of labile ligands from C-2, either through a dichlorination process or DMSO release, followed by ketone coordination to generate intermediate I. This step is presumably promoted by heating. Subsequently, intermediate I reacts with ethanol in the presence of a base to form a ruthenium-ethoxide complex II. This is followed by hydride addition to the β-position and β-hydride elimination, resulting in the release of acetaldehyde and the formation of the hydride complex III. Acetaldehyde is known to react with additional ethanol to form ethyl acetate and release hydrogen. Finally, the ruthenium complex III reacts with another ketone, releasing the secondary alcohol and regenerating the catalytically active ruthenium complex I.
Conclusions
In summary, two new ruthenium complexes with triazole-based ligands (C-1 and C-2) have been synthesized in a facile manner with good yields. Both complexes were fully characterized. Complex C-2, bearing the mer-tridentate ligand, proved to be air-, water-, and thermally stable, as well as a highly active Ru catalyst for the homogeneous hydrogenation of C=O bonds, selectively producing alcohols. It operated under relatively mild conditions and addressed a broad substrate scope, covering alkyl- and aryl-ketones as well as aryl-aldehydes. The transfer hydrogenation of ketones and aldehydes using ethanol as the source of hydrogen and solvent produced secondary and primary alcohols in good to excellent yields with high selectivity. The successful use of this Ru(II) species suggests its potential for other transformations. Efforts to further explore its catalytic activity in other hydrogenation reactions are currently underway in our laboratory, as well as the exploration of the potential biological activity of this compound, given the high-water solubility of C-2 and its biological components, such as triazole moieties. In addition, current investigations are focused on further mechanistic details. These results will be disclosed in due time.

