Articles
DFT Calculation, ADME/T and Molecular Docking Approach of Methyl 2-oxo-1,2-dihydrofuro[3,4-d] pyrimidine-3(4H)carboxylate

-
Publication dates-
March 04, 2025
Jul-Sep , 2024
- Article in PDF
- Article in XML
- Automatic translation
- Send this article by e-mail
- Share this article +
Abstract.
The optimized geometry of methyl 2-oxo-1,2-dihydrofuro[3,4-d] pyrimidine-3(4H) carboxylate (FP) was determined by density functional theory calculations. Geometric properties of FP such as bond length, bond angle, dihedral bond angle, and HOMO-LUMO energies in the gas phase were calculated by using the Gaussian program. Delocalization of the molecule’s charge was analyzed using Mulliken Population Analysis (MPA) and Natural Population Analysis (NPA) approaches. Electrophilic and nucleophilic regions of FP were identified by drawing a molecular electrostatic potential map. NMR and FTIR spectra were calculated with the B3LYP and 6-311++G (2d, p) basis set and a detailed FTIR analysis was performed by using the VEDA program. To determine the consistency of the calculated NMR and FTIR spectra, they were compared with their corresponding experimental NMR and FTIR spectra. Molecular insertion studies of FP with six different cancer proteins were analyzed and their interactions were evaluated. Data on the pharmacokinetics and drug affinity of FP were obtained through the Swiss ADME and ADMET programs.
Keywords::
Dihydrofuro [3,4-d], pyrimidine, DFT/B3LYP, molecular docking, swiss ADME, ADMET
Introduction
Pyrimidine, an aromatic heterocyclic compound containing nitrogen, is found in the structure of DNA and RNA, which are important for life. Synthesized pyrimidine-derived compounds can be used in many areas due to their features such as antifungal, anti-tumor, anti-inflammatory, anti-viral, anti-bacterial, anti-proliferative, anti-Alzheimer's, anti-tuberculosis, anti-β-glucuronidase, anti-HIV and diuretic [1-10]. Also, some cancer drugs like Xeloda [11], cytarabine [12] and gemcitabine [13], 5-fluorouracil [14] contain pyrimidine as the main component. Furopyrimidine compounds are synthesized by forming a pyrimidine ring next to the furan ring by various reactions. These compounds have biological properties similar to pyrimidines, which have been the subject of many studies in recent years [15-17]. It can be said that there is not much literature on quantum chemical calculations, molecular docking, ADME, and ADMET for furopyrimidine derivative compounds. For this purpose, methyl 2-oxo-1,2-dihydrofuro[3,4-d] pyrimidine-3(4H) carboxylate (FP) was first optimized using density functional theory (DFT). Bond angles, dihedral angles, bond lengths, dipole moments, molecular electrostatic potential map (MEP), highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) of FP were obtained with the Gaussian G09w package program [18]. Then, the calculated spectrum values of FTIR and NMR were compared with the experimental spectrum values reported by Yilmaz et al. [19]. Finally, the biopotential of FP was evaluated by molecular docking with six different cancer proteins. Data on the pharmacokinetics and drug affinity of FP were obtained through the Swiss ADME [20] and ADMET programs [21].
-
1Bioorg. Chem., 2021
-
10Bioorg. Med. Chem. Lett., 2019
-
11Nucl. Med. Biol., 2004
-
12Am. J. Hematol., 2012
-
13Cell Metab., 2019
-
14J. Chem. Thermodyn., 2021
-
15Bioorg. Chem., 2021
-
17Eur. J. Med. Chem., 2018
-
18Gaussian 09Revision B.01, 2009
-
19Tetrahedron., 2017
-
20Sci Rep., 2017
-
21Bioinformatics., 2018
Theorical method
Geometric calculations and molecular configuration of the FP were performed in the Gaussian G09w package program with the 6-311++G (2d, p) basis set using B3LYP [22-24]. MAP and FMO were also calculated by the same method. The gauge invariant atomic orbitals (GIAO) method was used for 1H and 13C NMR. Since the experimental data were obtained in dimethylsulphoxide (DMSO), theoretical calculations were made in DMSO for comparison, and the solvent effect on the chemical shift value was also investigated by calculating NMR in the gas phase. The calculated IR vibration frequencies are multiplied by the correction factor 0.9613 which provides the best agreement between the theoretical data and the experimental FTIR frequencies for B3LYP/6-31G(d) method [24]. A detailed potential energy distribution (PED) analysis was also performed using the VEDA program. The visualization of all data was carried out in GaussView 5.0 [25-27].
-
22Spectrochim. Acta A Mol. Biomol. Spectrosc., 2011
-
24Exploring Chemistry with Electronic Structure Methods, 1996
-
24Exploring Chemistry with Electronic Structure Methods, 1996
-
25Spectrochim. Acta A Mol. Biomol. Spectrosc., 2015
-
27Gauss ViewVersion 5, 2009
Molecular docking
Since many in vivo and in vitro experiments are required, it is very difficult to determine whether the synthesized compounds have biological effects. Molecular docking calculation of ligand-receptor interactions provides easier information about the bioactivity of the compound [28-31]. The best ligand-receptor interaction can be examined by choosing the lowest energy interaction among the molecular docking results. Molecular docking studies were performed with Auto Dock Tools 1. 5. 6. [32,33]. Molecular docking FP with various cancer proteins, namely brain cancer, breast cancer, gastric cancer, liver cancer, lung cancer, and skin cancer [34-39]. The 3D crystal structure of cancer proteins was taken from Protein Data Bank (Brain Cancer Protein PDB ID: 1QHT, [40] Breast Cancer Protein PDB ID:1JNX [41], Gastric Cancer Protein PDB ID:1BJ7 [42], Liver Cancer Protein PDB ID:3WZE [43], Lung Cancer Protein PDB ID:2ITO [44], Skin Cancer Protein PDB ID:2VCJ [45]. Water molecules were removed, and polar hydrogens and Kollman United charges were added as the biomolecules were prepared for docking. The FP molecule was prepared for docking by minimizing its energy at the Gaussian 09w program [18]. Partial Charles of FP was calculated by Geistenger Method. The spacing between grid points was 0.375 Å. Autodock docking studies were examined by using the Lamarckian Genetic Algorithm (LGA). As a result of the calculations, the lowest energy interaction was selected, and the results were interpreted. All FP-biomolecule interactions were illustrated using Pymol and Chimera software [46-48]. LigPlot+ v.1.4.5 software was used to show the H bonds in detail [49].
-
28Phytomed. Plus., 2022
-
31Drug Discovery Today, 2021
-
32J. Comput. Chem., 2010
-
33J. Comput. Chem., 2009
-
34Polycycl. Aromat. Compd., 2023
-
39J. Biomol. Struct. Dyn., 2021
-
40J. Mol. Biol., 2000
-
41Nat. Struct. Biol., 2001
-
42J. Biol. Chem., 1999
-
43ACS Med. Chem. Lett., 2015
-
44Cancer cell., 2007
-
45J. Med. Chem., 2008
-
18Gaussian 09Revision B.01, 2009
-
46Chemom. Intell. Lab. Syst., 2017
-
48J. Comput. Chem., 2004
-
49J. Chem. Inf. Model., 2011
Swiss ADME and ADMET Lab
It is important to evaluate the bioavailability of compounds using computer-based programs for the discovery of new drugs. Compounds can be classified as drug-like and non-drug-like according to the Lipinski rule [50]. Physicochemical and pharmacokinetic properties of FP were obtained using the Swiss ADME free website http://www.swissadme.ch/index.php (Swiss Institute of Bioinformatics, Switzerland) [20]. ADMET (absorption, distribution, metabolism, elimination, toxicity) is an important part of drug discovery and provides information about the drug's behaviour in metabolism. ADMET properties of FP were obtained using the online ADMET database, https://admetmesh.scbdd.com/service/screening/molecule [51].
-
50Drug. Discov. Today. Technol., 2004
-
20Sci Rep., 2017
-
51J. Chem. Inf. Model., 2012
Results and discussion
Molecular geometry
Molecular geometry is defined as a three-dimensional arrangement of atoms in a molecule. According to the differences in the positions and orientations of the atoms, a lot of conformations can be determined for a compound. Each different conformation affects the energy of the molecule, its physical and chemical properties, its reactivity, and its interaction with other molecules [52]. The most stable optimized geometry of the FP at its minimum energy level obtained by DFT calculations is shown in Fig. 1.
-
52Spectrochim. Acta A Mol. Biomol. Spectrosc., 2015
Thumbnail
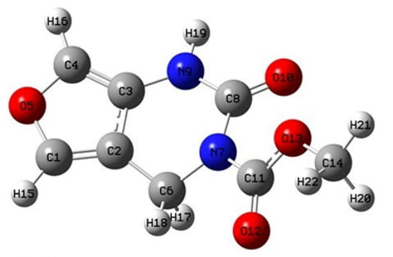
Fig. 1
Molecular configuration of FP.
Molecular configuration of FP.
The selected parameters for structural analysis, namely bond lengths, bond angles, and dihedral bond angles for the gas phase and in the DMSO solution of FP are given in Table 1. The Cartesian coordinates of the atoms in the minimized structure in the gas phase are given in supplementary S1. There are slight differences between the calculation in the gas phase and DMSO, except for dihedral angle H19-N9-C8-O10 as it turns to a positive value for the “in DMSO” case from its negative value for the gas phase. The dihedral angles (C6-C2-C3-N9) is are.1(1)° on average of the values obtained from both cases, while (C2-C6-N7-C8) is ~44.58(35)° exhibiting a bent pyrimidine ring. The flat dihedral angle (C1-C2-C3-N9) is ~180° showing that the (C6-C2-C3-N9) side of pyrimidine and furan rings are almost in the same plane while the angle ~162.89 (1,18)° of (N9-C8-N7-C11) indicates the separation of the FP tail from (C2-C6-N7-C8) plane of the pyrimidine ring.
Table 1
Theoretical bond lengths (Å), bond angles (◦), and dihedral bond angles (◦) of FP. Atom labels refer to Fig. 1.
Theoretical bond lengths (Å), bond angles (◦), and dihedral bond angles (◦) of FP. Atom labels refer to Fig. 1.
| Bond Length (Å) | Bond Angles (°) | Dihedral Bond Angles (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Gas | DMSO | Gas | DMSO | Gas | DMSO | |||
| C1-C2 | 1.354 | 1.354 | C1-C2-C3 | 105.97 | 106.06 | H16-C4-O5-C1 | -179.31 | -179.55 |
| C2-C3 | 1.423 | 1.423 | C3-C4-O5 | 108.75 | 108.53 | C1-C2-C3-C4 | -0.16 | -0.18 |
| C1-H15 | 1.076 | 1.076 | C3-C4-H16 | 134.58 | 134.48 | C2-C3-C4-O5 | -0.31 | -0.27 |
| C6-H18 | 1.086 | 1.085 | H16-C4-O5 | 116.65 | 116.99 | C4-C3-C2-C6 | 178.21 | 178.03 |
| C14-H20 | 1.089 | 1.088 | C4-O5-C1 | 107.53 | 107.68 | C6-C2-C3-N9 | -1.98 | -2.19 |
| C4-O5 | 1.371 | 1.373 | O5-C1-C2 | 116.17 | 109.99 | H19-N9-C8-O10 | -1.34 | 0.23 |
| C1-O5 | 1.364 | 1.364 | N7-C11-O12 | 122.17 | 122.19 | N9-C8-N7-C11 | -164.08 | -161.71 |
| C8-O10 | 1.209 | 1.218 | C11-O13-C14 | 115.19 | 115.61 | N9-C8-N7-C6 | 28.54 | 29.15 |
| C11-O12 | 1.210 | 1.215 | C2-C6-N7 | 108.62 | 108.49 | O10-C8-N7-C11 | 16.69 | 19.05 |
| C14-O13 | 1.438 | 1.444 | C6-N7-C8 | 120.75 | 120.65 | C8-N7-C11-O12 | -158.54 | -158.83 |
| C6-N7 | 1.484 | 1.488 | N7-C8-N9 | 114.16 | 114.78 | N7-C11-O13-C14 | -178.25 | -179.53 |
| C8-N7 | 1.420 | 1.419 | H19-N9-C3 | 121.91 | 121.24 | C11-O13-C14-H21 | 177.59 | 178.19 |
| C8-N9 | 1.382 | 1.370 | N9-C8-O10 | 121.14 | 121.29 | O12-C11-O13-C14 | 5.13 | 3.76 |
| C3-N9 | 1.389 | 1.391 | O10-C8-N7 | 124.69 | 123.92 | C1-C2-C3-N9 | 179.63 | 179.60 |
| C11-N7 | 1.399 | 1.395 | C8-N7-C11 | 122.81 | 122.89 | C2-C6-N7-C8 | -44.95 | -44.22 |
| N9-H19 | 1.009 | 1.009 | O12-C11-O13 | 124.75 | 124.35 | C6-C2-C3-N9 | -1.99 | -2.19 |
The charge distribution in the atoms in the molecule is important for determining the electrostatic interaction and molecular force fields [53,54]. The atomic charge distributions of the FP are obtained by Mulliken Population Analysis (MPA), and Natural Population Analysis (NPA) methods within the framework Gaussian 09, which are listed in Table 2. There are differences when comparing the calculated atomic charges in the two methods. The charges such as C1, C2, and C3 are given discrepant by the two methods. MPA(C1) is -0.116 and NPA(C1) is 0.154 with a change in the sign. C2 has the same issue. Other atoms, even though they show the same sign, they differ in magnitude; for example, MPA(C3) is 0.312, and NPA(C3) is 0.078. However, the two methods provide good agreement on the charges of some atoms, such as O5, C4, C11, and H16. Overall, the oxygen and nitrogen atoms in the molecule are negatively charged, while the hydrogen atoms are positively charged, as usual. Amongst the carbon atoms, the highest negative charge is located on C14, while the highest positive charge is located on C11. The carbon atoms have negative or positive charges with respect to the electronegative atoms around them.
-
53J. Mol. Struct., 2015
-
54J. Mol. Struct., 2021
Table 2
Atomic charges of FP compound by Mulliken Population Analysis (MPA), and Natural Population Analysis (NPA) methods. Labels refer to Fig. 1.
Atomic charges of FP compound by Mulliken Population Analysis (MPA), and Natural Population Analysis (NPA) methods. Labels refer to Fig. 1.
| Atom | MPA | NPA | Atom | MPA | NPA |
|---|---|---|---|---|---|
| C1 | -0.116 | 0.154 | O13 | -0.318 | -0.533 |
| C2 | 0.549 | -0.147 | N7 | -0.343 | -0.564 |
| C3 | 0.312 | 0.078 | N9 | -0.509 | -0.616 |
| C4 | 0.055 | 0.082 | H15 | 0.181 | 0.200 |
| C6 | -0.963 | -0.176 | H16 | 0.180 | 0.200 |
| C8 | 0.559 | 0.843 | H17 | 0.161 | 0.205 |
| C11 | 0.713 | 0.968 | H18 | 0.212 | 0.244 |
| C14 | -0.121 | -0.200 | H19 | 0.275 | 0.412 |
| O5 | -0.366 | -0.455 | H20 | 0.164 | 0.186 |
| O10 | -0.458 | -0.608 | H21 | 0.145 | 0.187 |
| O12 | -0.468 | -0.644 | H22 | 0.157 | 0.184 |
The MPA and NPA methods obtain the results sensitively depending on their basis sets which induce change in the calculated net charges [54]. However, the results given by NPA are supposed to be more reliable as their calculations are based on the natural charges [55].
-
54J. Mol. Struct., 2021
-
55J. Mol. Struct., 2020
Molecular orbital energy analysis and electronic properties
Quantum chemical parameters were calculated with FMO analysis, and the highest occupied molecular orbital energy (EHOMO), the lowest unoccupied molecular orbital energy (ELUMO) together with the energy gap (∆E=ELUMO-EHOMO) given in Fig.2. ∆E determines the molecular properties, namely softness and chemical reactivity [54,56-58]. In this respect, the ∆E energy difference of the FP calculated using the B3LYP and the 6-311++G (2d, p) basis set was 5.81 eV as a single molecule ( 5.64 eV in DMSO) related to the dipole moments of FP molecule which were found μgas=1,92 Debye and μDMSO=2,99 Debye in the gas phase and iMSO, respectively. The dipole moments were obtained by the conformal minimization of the molecule. These values make FP a high enough polarized pyrimidine molecule as compared to that of 4.86 eV for 5-Fluorouracil, a commercial anticancer reagent [59].
-
54J. Mol. Struct., 2021
-
56J. Mol. Struct., 2022
-
58Chem. Pap., 2020
-
59Spectrochim. Acta A Mol. Biomol. Spectrosc., 2017
Thumbnail
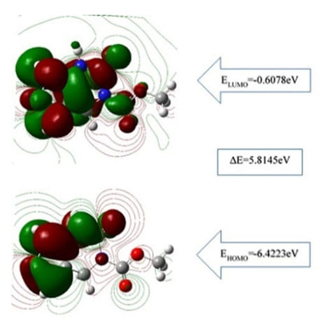
Fig. 2
EHOMO and ELUMO energy levels for FP.
EHOMO and ELUMO energy levels for FP.
The calculations of EHOMO and ELUMO energy levels lead to automatic determination of ionization energy (I) and electron affinity (A) which are expressed as
e1
I
=
-
E
H
O
M
O
(1)
and
e2
A
=
-
E
L
U
M
O
(2)
As EHOMO indicates the electron-donating ability while ELUMO refers to the electron-accepting ability. Once reaching the concerning parameters above, some characterizing parameters such as electronegativity (X), chemical hardness (η), and softness (σ) from the following relations:
e3
χ
=
I
+
A
2
(3)
e4
η
=
I
-
A
2
(4)
and
e5
σ
=
1
η
(5)
The molecular electronic properties of FP obtained are given in Table 3. The high electronegativity values χgas=3.51 eV and χDMSO=3.53 eV indicates the strong attraction to covalent bonds [34]. Also, the large chemical hardness ηgas=2.91 eV and ηDMSO=2.82 eV indicates chemical stability, and the molecule can be considered as nontoxic because of the low softness values σgas=0.34 eV-1 and σDMSO=0.35 eV-1.
-
34Polycycl. Aromat. Compd., 2023
Table 3
Electronic structure values of FP.
Electronic structure values of FP.
| Phase | EHOMO (eV) | ELUMO (eV) | ΔE (eV) | I(eV) | A(eV) | χ(eV) | η(eV) | σ (eV-1) |
|---|---|---|---|---|---|---|---|---|
| gas | -6.42 | -0.61 | 5.81 | 6.42 | 0.61 | 3.51 | 2.91 | 0.34 |
| DMSO | -6.36 | -0.71 | 5.64 | 6.36 | 0.71 | 3.53 | 2.82 | 0.35 |
Molecular electrostatic potential
The molecular electrostatic potential of FP was calculated and shown in Fig. 3. It shows the positively charged parts of the molecule in blue colour and the negatively charged parts in red colour [60-62].
-
60J. Mol. Struct., 2021
-
62J. Mol. Struct., 2022
Thumbnail
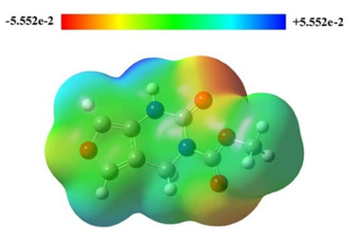
Fig. 3
Molecular electrostatic potential for FP.
Molecular electrostatic potential for FP.
As MEP allows us to have insight into the chemical reactivity of a molecule, the highest negative charge -0.059 region by red shade in Fig.3 is the electrophilic region. The negative electrostatic potential indicates the concentrated electron density in the molecule interacting with the receptors and forming hydrogen bonds with the positive sites of amino acids. They correspond to the tops of two carbonyls. The highest positive charge +0.059 region by blue shade is the nucleophilic region which corresponds to the hydrogen in the N-H bond. The green area can be considered almost a neutral region.
Experimental and theoretical 13C NMR and 1H NMR chemical shift values
Experimental and theoretical 1H NMR and 13C NMR chemical shift values of FP are listed in Table 4 according to TMS δ/ppm. The experimental NMR values were taken in DMSO [19], and theoretical NMR data were calculated in the gas phase and DMSO phase. In the FP structure, there are five different hydrogens attached to aliphatic and aromatic carbons and amine-bound, and all H peaks were observed as a singlet. In addition, eight different carbon peaks were found in the aliphatic, aromatic, and carbonyl structures.
-
19Tetrahedron., 2017
When discussing the 1H NMR spectra of the FP, the experimental chemical shift values of the protons-bounded aliphatic carbons were observed as a singlet in a value, while the theoretical chemical shift values were seen as two different peaks. The experimental value of the ring protons, CH2 was at 4.71 ppm as a singlet, while the theoretical values of H17 and H18 were 4.50 and 5.19 ppm, respectively. Ester protons, CH3 experimental value was in the 3.78 ppm as a singlet, while the theoretical values of H21 and H20, H22 were in 3.72 and 3.87 ppm, respectively. The experimental value of amine proton was 9.96 ppm as a broad singlet, whereas the theoretical value of H19 was 6.42 ppm. The experimental values of aromatic ring protons were at 7.27 and 7.46 ppm, whereas the theoretical values of H15 and H16 were at 7.31 and 7.37 ppm, respectively.
Table 4
The experimental and theoretical 1H NMR and 13C NMR chemical shift values of FP according to TMS δ/ppm. (s.: singlet, br.: broad Assign.: assignments) The hydrogen and carbon labels can be followed in Fig. 1.
The experimental and theoretical 1H NMR and 13C NMR chemical shift values of FP according to TMS δ/ppm. (s.: singlet, br.: broad Assign.: assignments) The hydrogen and carbon labels can be followed in Fig. 1.
| 1 H NMR | ||||
|---|---|---|---|---|
| Experimental | Theoretical | |||
| Assign. | δ(ppm) | Assign. | Gas phase | DMSO |
| CH3 | 3,73 (s) | H21 | 3.56 | 3.72 |
| H20, H22 | 3.83 | 3.87 | ||
| CH2 | 4.71 (s) | H17 | 4.34 | 4.50 |
| H18 | 5.14 | 5.19 | ||
| NH | 9.96 (br. s) | H19 | 5.75 | 6.42 |
| CH (arom.) | 7.27 (s) | H15 | 7.05 | 7.31 |
| CH (arom.) | 7.46 (s) | H16 | 7.12 | 7.37 |
| 13 C NMR | ||||
| Experimental | Theoretical | |||
| Assign. | δ(ppm) | Assign. | Gas phase | DMSO |
| CH2 | 29.74 | C6 | 43.24 | 44.06 |
| CH3 | 54.28 | C14 | 54.87 | 55.95 |
| C(arom.) | 111.13 | C2 | 117.37 | 117.28 |
| CH(arom.) | 124.88 | C4 | 126.54 | 129.19 |
| C(arom.) | 126.17 | C3 | 132.70 | 131.85 |
| CH(arom.) | 136.96 | C1 | 140.50 | 141.87 |
| C=O | 150.05 | C8 | 153.01 | 156.06 |
| C=O | 155.32 | C11 | 162.07 | 163.40 |
Considering the 13C NMR spectra of the FP, the experimental values of the aliphatic ring (CH2) and ester methyl carbon peaks (CH3) were at 29.74 and 54.28 ppm, whereas the theoretical values of C6 and C14 were at 44.06 and 55.95 ppm, respectively. The aromatic carbons’ experimental values were 111.13, 124.88, 126.17, and 136.96 ppm, whereas theoretical values of C2, C4, C3, and C1 were 117.28, 129.18, 131.85, and 141.87, respectively. The experimental values of ring carbonyl carbon and aliphatic carbonyl carbon were at 150.05 and 155.32 ppm, while theoretical values of C8 and C11 were at 156.06 and 163.40 ppm, respectively. According to the NMR results, the experimental and theoretical chemical shift values are compatible. The theoretical and experimental NMR spectra were given in the supplementary (S2-S5).
Experimental and theoretical vibration frequencies
The molecular structure of FP contains twenty-two atoms, which gives forty-four normal modes of vibrations. While experimental vibration values were taken from the literature [19], theoretical vibration values were calculated using the DFT/B3LYP method and 6-311++G (2d, p) basis set. The FTIR spectrum was analyzed based on characteristic peaks such as amine, aromatic, carbonyl, and CH2 vibrations. The experimental and theoretical values of the vibrational wavenumber of FP are shown in Fig. 4. For analysis, all the calculated FTIR, compared to experimental values, and details of the percentage of potential energy distribution (PED) assignment are listed in Table 5. When performing PED assignments, it was found that there were thirty-four stretching vibrations, thirty-six in-plane bending vibrations, four out of plane bending vibrations, and eighteen torsional vibrations. When the experimental and theoretical values were compared, they were found to be well-matched.
-
19Tetrahedron., 2017
Thumbnail
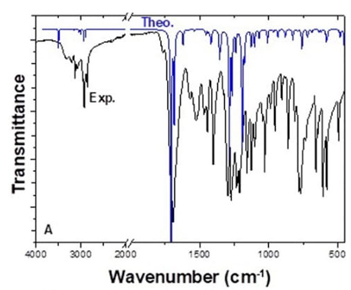
Fig. 4
Experimental and theoretical FTIR spectrum of FP (cm-1).
Experimental and theoretical FTIR spectrum of FP (cm-1).
Table 5
Experimental and theoretical FTIR values of FP (cm-1).
Experimental and theoretical FTIR values of FP (cm-1).
| Experimental | Theoretical | Vibration assignment (PED) | ||
|---|---|---|---|---|
| No | Unscaled | Scaled | ||
| 1 | 3321 | 3633 | 3492 | νN9-H19 (100) |
| 2 | 3071 | 3282 | 3155 | νC4-H16 (95) |
| 3 | 3048 | 3271 | 3144 | νC1-H15 (95) |
| 4 | 3158 | 3036 | νC14-H21 (80), νC14-H20 (19), νC14-H22 (19) | |
| 5 | 3137 | 3016 | νC6-H18 (98) | |
| 6 | 2980 | 3127 | 3006 | νC14-H20, νC14-H22 (100) |
| 7 | 3054 | 2936 | νC14-H20, νC14-H21 (80) | |
| 8 | 2925 | 3010 | 2894 | νC6-H17 (98) |
| 9 | 1694 | 1777 | 1708 | νC8-O10 (77) |
| 10 | 1686 | 1752 | 1684 | νC11-O12 (81) |
| 11 | 1571 | 1686 | 1621 | νC1-C2, νC3-C4 (76) |
| 12 | 1528 | 1599 | 1537 | νC1-C2 (52), νC3-N9 (52) δH15-C1-O5 (12), δH16-C4-O5 (12), δC1-O5-C4 (11), δC3-C4-O5 (11) |
| 13 | 1465 | 1513 | 1454 | δH18-C6-H17 (74), τH18-C6-C2-C1 (12) |
| 14 | 1458 | 1497 | 1439 | δH20-C14-H22 (70), τH20-C14-O13-C11 (21), τH22-C14-O13-C11-(21) |
| 15 | 1440 | 1485 | 1428 | δH21-C14-H20 (38), δH22-C14-H21 (35), τH21-C14-O13-C11 (15), τH20-C14-O13-C11 (10), τH22-C14-O13-C11 (10) |
| 16 | 1406 | 1469 | 1412 | δH19-N9-C3, δC2-C1-O5 (44) |
| 17 | 1326 | 1411 | 1356 | δC6-N7-C8 (14) |
| 18 | 1301 | 1402 | 1348 | νC3-C4, νC3-N9 (24) |
| 19 | 1293 | 1337 | 1285 | νC11-N7 (44), νC11-O13 (44), δH17-C6-C2 (10) |
| 20 | 1270 | 1289 | 1239 | νC8-N9 (10) |
| 21 | 1234 | 1244 | 1196 | δH16-C4-O5, δH15-C1-O5 (67) |
| 22 | 1205 | 1224 | 1177 | δC8-N7-C11, δN9-C8-O10, δC3-N9-C8, δO12-C11-O13, δC3-C4-O5 (15) |
| 23 | 1159 | 1172 | 1127 | νC1-O5, νC6-N7 (54) |
| 24 | 1128 | 1147 | 1103 | νC1-O5, νC4-O5, νC6-N7 (61) |
| 25 | 1100 | 1112 | 1069 | νC11-N7 (22), νC11-O13 (22), νC14-O13 (11) |
| 26 | 1040 | 1049 | 1008 | νC1-O5, νC4-O5 (64) |
| 27 | 985 | 1005 | 966 | τH17-C6-C2-C1 (26) |
| 28 | 951 | 916 | 881 | δC1-O5-C4, δN7-C8-N9 (44) |
| 29 | 902 | 858 | 825 | δC3-C4-O5, δN9-C8-O10, δN7-C8-N9 (76) |
| 30 | 859 | 822 | 790 | δO12-C11-O13, δC11-O13-C14, δC3-N9-C8 (41) |
| 31 | 781 | 790 | 759 | τH15-C1-O5-C4 (57), τC1-O5-C4-C3 (11), γO12-N7-O13-C11 (66) |
| 32 | 766 | 749 | 720 | γO10-N9-N7-C8 (85) |
| 33 | 724 | 743 | 714 | νC8-N7 (12), τC6-N7-C8-N9 (10) |
| 34 | 660 | 715 | 687 | τH16-C4-O5-C1 (45), τC2-C1-O5-C4 (18), τC1-O5-C4-C3 (18) |
| 35 | 608 | 606 | 583 | δC3-N9-C8, δO12-C11-O13, δN9-C8-O10 (35) |
| 36 | 580 | 502 | 483 | τH19-N9-C3-C2 (30) |
| 37 | 489 | 435 | 418 | δN7-C11-O13, δN9-C8-O10,-δO12-C11-O13, δC8-N7-C11 (49) |
| 38 | 334 | 321 | γN9-C2-C4-C3 (23) | |
| 39 | 324 | 311 | δC11-O13-C14, δC8-N7-C11 (60) | |
| 40 | 296 | 285 | τN7-C8-N9-C3 (10) | |
| 41 | 257 | 247 | δC8-N7-C11, δC11-O13-C14, δN7-C11-O13, δN7-C8-N9 (10) | |
| 42 | 222 | 213 | τC8-N9-C3-C2 (34), τC14-O13-C11-N7 (11) | |
| 43 | 169 | 162 | γC11-C6-C8-N7 (17) | |
| 44 | 62 | 60 | τO13-C11-N7-C6 (71) | |
NH vibrations
The stretching vibrations of the amide NH bond were observed around 3300-3600 cm-1 [62,63]. In FP, N9-H19 stretching vibration was theoretically observed at 3492 cm-1 with PED contribution is 100 % and experimentally at 3321 cm-1 as a strong peak. Furthermore, H19-N9-C3 in-plane bending vibration was observed theoretically at 1412 cm-1 and experimentally at 1406 cm-1. The H19-N9-C3-C2 torsional vibration was observed theoretically at 483 cm-1 and experimentally at 580 cm-1.
-
62J. Mol. Struct., 2022
-
63Spectrochim. Acta A. Mol. Biomol. Spectrosc., 2018
Aromatic CH vibrations
Aromatic CH bond stretching vibrations are usually just over 3000 cm-1 [63,64]. There are two different aromatic CH in the FP structure, and their vibrational values were very close. C4-H16 and C1-H15 stretching vibrations were theoretically observed at 3144 cm-1 and 3157 cm-1, and experimentally at 3193 cm-1 and 3139 cm-1, respectively. H16-C4-O5 and H15-C1-O5 in-plane bending vibrations had the same value and two vibrations; theoretically observed at 1537 cm-1 and 1196 cm-1, experimentally at 1528 cm-1 and 1234 cm-1. Moreover, H15-C1-O5-C4 and H16-C4-O5-C1 torsional vibrations were observed theoretically at 759 cm-1 and 687 cm-1 experimentally at 781 cm-1 and 660 cm-1, respectively.
-
63Spectrochim. Acta A. Mol. Biomol. Spectrosc., 2018
-
64J. Mol. Struct., 2022
Carbonyl vibrations
Depending on the electronegative atoms attached to the carbonyl group, the carbonyl group gives a stretching vibration in the range of 1650-1800 cm-1 [62,63]. There are two different carbonyl groups located between nitrogen-nitrogen and oxygen-nitrogen in the FP structure. The carbonyl group (C8-O10) located between nitrogen and nitrogen in the ring structure gave a higher frequency peak than the other (C11-O12). While C8-O10 and C11-O12 stretching frequency were observed theoretically at 1708 and 1684 cm-1, experimentally at 1694 and 1686 cm-1, respectively. In addition, N9-C8-O10 in-plane bending vibrations were theoretically observed at 1177, 825, 583, 418 cm-1, and experimentally at 1205, 902, 608, 489 cm-1.
-
62J. Mol. Struct., 2022
-
63Spectrochim. Acta A. Mol. Biomol. Spectrosc., 2018
CH2 vibrations
The characteristic vibration of the CH2 is the stretching vibration in the range of 2900-3000 cm-1 and the in-plane bending vibration around 1450 cm-1 [65]. The C6-H18 and C6-H17 stretching vibrations were observed theoretically at 3016 and 2894 cm-1, respectively, and experimentally at 2925 cm-1. H18-C6-H17 in-plane bending vibration was observed theoretically at 1454 cm-1 and experimentally at 1465 cm-1.
-
65Spectrochim. Acta A Mol. Biomol. Spectrosc., 2012
Molecular docking
Molecular docking calculations are used to gain information about ligand-receptor interactions and play an important role in drug discovery. As a result of these calculations, it can be found out which region of the protein the ligand binds to, what kind of interactions it makes, and their binding energies [54,60,66,67]. In this study, molecular docking calculations with the brain (PDB ID: 1QHT), breast (PDB ID: 1JNX), gastric (PDB ID: 1BJ7), liver (PDB ID: 3WZE), lung (PDB ID: 2ITO), and skin (PDB ID: 2VCJ) cancer proteins to see the biological potential of FP were examined. Many docking studies were carried out and the lowest energies interaction was selected. The calculated lowest binding energies of the target biomolecules and FP are given in Table 6. In addition, FP makes more stable interactions with brain and liver cancer cells than others. The binding sites of FP with target biomolecules are shown in Figure 5. Looking at the structure of the FP compound, it is seen that it is an active compound-containing pyrimidine ring, furan ring, and ester. When calculated results are examined, FP makes hydrophobic interactions with ARG130, ARG132, TRP228, ARG236, ILE238, MET240, ARG292 amino acids in brain cancer protein, SER1755, ARG1758, LYS1759, ILE1760, ARG1762, TYR1845, GLN1846, CYS1847 amino acids in breast cancer protein, ARG40, ILE149, ASP152, ASN153, CYS154, PRO156 amino acids in gastric cancer protein, HIS816, LYS868, ALA881, SER884, GLU885, ILE888, ARG1027, ASP1046, GLY1048, LEU1049, BAX1201 amino acids in liver cancer protein, SER 719, ALA722, PHE723, VAL726, LYS745, ARG841, LEU844, ASP855, IRE2020 amino acids in lung cancer protein, ARG46, GLU47, SER50, ASN51, GLY132, GLN133, GLY135, GLY137 amino acids in skin cancer protein.
-
54J. Mol. Struct., 2021
-
60J. Mol. Struct., 2021
-
66Spectrochim. Acta. A Mol. Biomol. Spectrosc., 2022
-
67Heliyon, 2019
Table 6
The binding energies of FP and target biomolecules.
The binding energies of FP and target biomolecules.
| Protein (PDB ID) | Types of cancer | Binding energy (kcal/mol) |
|---|---|---|
| 1QH4 | Brain cancer | -6.1 |
| 1JNX | Breast cancer | -4.9 |
| 1BJ7 | Gastric cancer | -4.4 |
| 3WZE | Liver cancer | -6.1 |
| 2ITO | Lung cancer | -5.9 |
| 2VCJ | Skin cancer | -4.8 |
Thumbnail
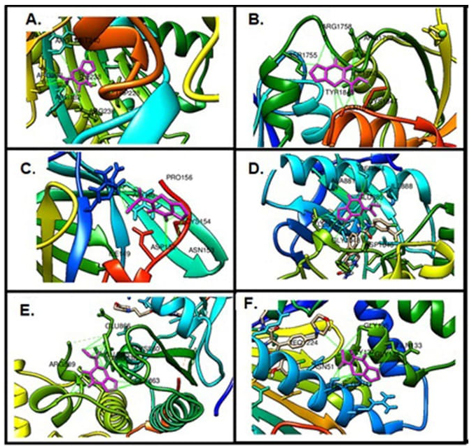
Fig. 5
The binding site of FP on target proteins, (A) Brain cancer protein, (B) Breast cancer protein, (C) Gastric cancer protein, (D) Liver cancer protein, (E) Lung cancer protein, (F) Skin cancer protein.
The binding site of FP on target proteins, (A) Brain cancer protein, (B) Breast cancer protein, (C) Gastric cancer protein, (D) Liver cancer protein, (E) Lung cancer protein, (F) Skin cancer protein.
H bonds are strong interactions that hold molecules together and the number of the H bonds gives information about the strength of the interaction. The H bond interactions between FP and targeted biomolecules are given in Fig. 6. Also, the number of H bonds made by FP with each biomolecule and the atoms forming the H bond are listed in Table 7.
Thumbnail
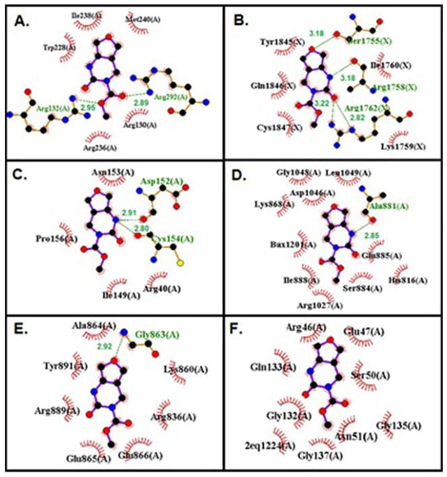
Fig. 6
The H bond interactions between FP and target proteins, (A) Brain cancer protein, (B) Breast cancer protein, (C) Gastric cancer protein, (D) Liver cancer protein, (E) Lung cancer protein, (F) Skin cancer protein.
The H bond interactions between FP and target proteins, (A) Brain cancer protein, (B) Breast cancer protein, (C) Gastric cancer protein, (D) Liver cancer protein, (E) Lung cancer protein, (F) Skin cancer protein.
Table 7
Summary of hydrogen bonding of furopyrimidine molecule with different types of biomolecule targets.
Summary of hydrogen bonding of furopyrimidine molecule with different types of biomolecule targets.
| Protein (PDB ID) | Number of H bond | Bonded residues | Bond distance (A◦) | |
|---|---|---|---|---|
| 1QH4 | 2 | Protein ARG292:HN | Ligand: O12 | 2.89 |
| Protein ARG132:HN | Ligand:O13 | 2.95 | ||
| 1JNX | 4 |
Protein SER1755:OH Protein ARG1758:O Protein ARG1762:HN Protein ARG1762:HN |
Ligand: O5 Ligand: N9H19 Ligand: O12 Ligand: O12 |
3.18 3.18 2.82 3.22 |
| 1BJ7 | 2 |
Protein ASP152:O Protein CYS154:O |
Ligand: N9H19 Ligand: N9H19 |
2.91 2.80 |
| 3WZE | 1 | Protein ALA881:O | Ligand: N9H19 | 2.85 |
| 2ITO | 1 | Protein LYS745:NH | Ligand: O12 | 3.21 |
| 2VCJ | - | - | - | - |
When it is examined in this study, FP makes two H bonds with ARG 132, ARG 292 amino acids in brain cancer protein, it makes four H bonds with SER1755, ARG1758, ARG1752 amino acids in breast cancer protein, it makes two H bonds with ASP152, CYS 154 amino acids in gastric cancer protein, it makes one H bond with ALA881 amino acid liver cancer protein, it makes one H bond with GLY863 amino acid in liver cancer protein, it doesn’t make H bond with an amino acid in skin cancer protein, and it makes two H bond with DA5.
When the H bonds were examined, it was seen that N9 in the FP structure is the H bond donor, and O5, O12, and O13 are H bond acceptors. It can be said that N9 and O12 atoms in the FP structure are the most active ones in forming H bonds.
Evaluation of pharmacokinetics and drug-likeness properties of FP
By evaluating the pharmacokinetic properties, toxicity, and bioavailability of the compounds, their usage or development as a drug can be achieved [68-70]. As shown in Table 8, FP follows the Lipinski Rule, Pfizer Rule, and GlaxoSmithKline Rule as a drug candidate. According to the ADME result, FP has a high GI (gastrointestinal) absorption value, which means that the drug is rapidly absorbed.
-
68J. Chem. Inf. Model., 2014
-
70Adv. Drug Delivery Rev., 2002
The molecular weight of FP being 196.16 g/mol and having 2 rotatable bonds indicate the conformity of the molecule. The number of hydrogen bond acceptors and donors is within the expected range, which indicates that they can interact strongly enough. Log P is a measure of the hydrophilicity and hydrophobicity of a molecule. The log P value of FP is compatible with the Lipinski rule. The topological polar surface area (TPSA) value of FP 71.78Å2 indicates that they have good permeability during the cellular plasma membrane and blood-brain barrier.
Table 8
Data of Lipinski rule, Pharmacokinetics, and Drug likeness.
Data of Lipinski rule, Pharmacokinetics, and Drug likeness.
| Mw | NBR | HBA | HBD | TPSA/A2 (≤140) | Consensus Log Po/w | Bioavailability Score | GI abs. |
|---|---|---|---|---|---|---|---|
| 196,16 | 2 | 4 | 1 | 71.78 | 0.21 | 0.55 | high |
The ADMET data of the FP which is computationally forecasted from its given molecular structure are shown in Table 9. It is exhibited that the FP has oral bioavailability, and is rapidly absorbed in the human intestine. Also, FP does not show skin sensitivities, AMES toxicity, and respiratory toxicity. The FP is not a P-glycoprotein inhibitor or substrate. Since P-glycoprotein pumps drugs back into the lumen, reducing drug absorption, it can be said that the bioavailability and bioactivity of the FP are high. A summary of the ADMET properties of FP is shown in Fig. 7.
Thumbnail
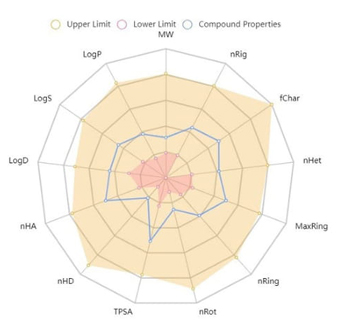
Fig. 7
Radar chart involved in ADMET properties of FP.
Radar chart involved in ADMET properties of FP.
Table 9
Pharmacokinetics and ADMET Data.
Pharmacokinetics and ADMET Data.
| Lipinski Rule | yes | Respiratory toxicity | no |
| Pfizer Rule | yes | P-glycoprotein inhibitior | no |
| GlaxoSmithKline Rule | yes | P-glycoprotein substrate | no |
| Human Intestinal Absorption | high | Solubility | yes |
| AMES toxicity | no | hERG (the human Ether-à-go-go-Related Gene) inhibitor | no |
| Skin sensitization | no |
Conclusions
In this study, the FP geometry was optimized using the B3LYP/6-311G++ (2d, p) method within density functional theory. The bond lengths, bond angles, dihedral angles, MEP, dipole moments, HOMO, and LUMO of FP were obtained with the Gaussian G09w program. Theoretical FTIR and NMR spectrum values were found to be compatible with the experimental spectrum values. Due to the wide biological activities of pyrimidine compounds, molecular docking studies of FP were carried out with various biomolecules. As a result of molecular docking studies, it was determined that N9 and O12 atoms of FP were the most active ones, and they make hydrogen bonds and hydrophobic interactions with biomolecules. Data on the pharmacokinetic and drug-likeness of FP was observed. For future studies, the synthesis of new dihydrofuro [3,4-d] pyrimidine derivatives may be desirable and could be a source of similar studies to determine their mechanisms as potential anticancer agents.
References
-
1Abdel-Aziz, S. A.; Taher, E. S.; Lan, P.; Asaad, G. F.; Gomaa, H. A.; El-Koussi, N. A.; Youssif, B. G. Bioorg. Chem. 2021, 111, 104890. DOI: https://doi.org/10.1016/j.bioorg.2021.104890. Links
-
2Aksinenko, A. Y.; Goreva, T. V.; Epishina, T. A.; Trepalin, S. V.; Sokolov, V. B. J. Fluor. Chem. 2016, 188, 191-195. DOI: https://doi.org/10.1016/j.jfluchem.2016.06.019. Links
-
3Ballesteros-Casallas, A.; Paulino, M.; Vidossich, P.; Melo, C.; Jiménez, E.; Castillo, J.-C.; Portilla, J.; Miscione, G. P. Eur. J. Med. Chem. 2022, 4, 100028. DOI: https://doi.org/10.1016/j.ejmcr.2021.100028. Links
-
4Basyouni, W. M.; Abbas, S. Y.; El‐Bayouki, K. A.; Dawood, R. M.; El Awady, M. K.; Abdelhafez, T. H. J. Heterocycl. Chem. 2021, 58, 1766-1774. DOI: https://doi.org/10.1002/jhet.4307. Links
-
5Elkanzi, N. A. A. Orient. J. Chem. 2020, 36, 1001-1015. DOI: http://dx.doi.org/10.13005/ojc/360602. Links
-
6Ali, F.; Khan, K. M.; Salar, U.; Iqbal, S.; Taha, M.; Ismail, N. H.; Perveen, S.; Wadood, A.; Ghufran, M.; Ali, B. Bioorg. Med. Chem. 2016, 24, 3624-3635. DOI: https://doi.org/10.1016/j.bmc.2016.06.002. Links
-
7Katariya, K. D.; Reddy, D. V. J. Mol. Struct. 2022, 1253, 132240. DOI: https://doi.org/10.1016/j.molstruc.2021.132240. Links
-
8Lamie, P. F.; Philoppes, J. N. Bioorg. Chem. 2021, 116, 105335. DOI: https://doi.org/10.1016/j.bioorg.2021.105335. Links
-
9Manzoor, S.; Prajapati, S. K.; Majumdar, S.; Raza, M. K.; Gabr, M. T.; Kumar, S.; Pal, K.; Rashid, H.; Kumar, S.; Krishnamurthy, S. Eur. J. Med. Chem. 2021, 215, 113224. DOI: https://doi.org/10.1016/j.ejmech.2021.113224. Links
-
10Raju, K. S.; AnkiReddy, S.; Sabitha, G.; Krishna, V. S.; Sriram, D.; Reddy, K. B.; Sagurthi, S. R. Bioorg. Med. Chem. Lett. 2019, 29, 284-290. DOI: https://doi.org/10.1016/j.bmcl.2018.11.036. Links
-
11Fei, X.; Wang, J.-Q.; Miller, K. D.; Sledge, G. W.; Hutchins, G. D.; Zheng, Q.-H. Nucl. Med. Biol. 2004, 31, 1033-1041. DOI: https://doi.org/10.1016/j.nucmedbio.2004.02.006. Links
-
12Scappini, B.; Gianfaldoni, G.; Caracciolo, F.; Mannelli, F.; Biagiotti, C.; Romani, C.; Pogliani, E. M.; Simonetti, F.; Borin, L.; Fanci, R. Am. J. Hematol. 2012, 87, 1047-1051. DOI: https://doi.org/10.1002/ajh.23308. Links
-
13Halbrook, C. J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P. Cell Metab. 2019, 29, 1390-1399. e6. DOI: https://doi.org/10.1016/j.cmet.2019.02.001. Links
-
14Verissimo, L. M.; Cabral, I.; Cabral, A. M.; Utzeri, G.; Veiga, F. J.; Valente, A. J.; Ribeiro, A. C. J. Chem. Thermodyn. 2021, 161, 106533. DOI: https://doi.org/10.1016/j.jct.2021.106533. Links
-
15Abd El-Mageed, M. M.; Eissa, A. A.; Farag, A. E.-S.; Osman, E. E. A. Bioorg. Chem. 2021, 116, 105336. DOI: https://doi.org/10.1016/j.bioorg.2021.105336. Links
-
16Gregorić, T.; Sedić, M.; Grbčić, P.; Paravić, A. T.; Pavelić, S. K.; Cetina, M.; Vianello, R.; Raić-Malić, S. Eur. J. Med. Chem. 2017, 125, 1247-1267. DOI: https://doi.org/10.1016/j.ejmech.2016.11.028. Links
-
17Hossam, M.; Lasheen, D. S.; Ismail, N. S.; Esmat, A.; Mansour, A. M.; Singab, A. N. B.; Abouzid, K. A. Eur. J. Med. Chem. 2018, 144, 330-348. DOI: https://doi.org/10.1016/j.ejmech.2017.12.022. Links
-
18Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J., Gaussian 09, Revision B.01, Gaussian Inc., Wallingford CT, 2009. Links
-
19Yılmaz, A. Ş.; Kaçan, M. Tetrahedron. 2017, 73, 4509-4512. DOI: https://doi.org/10.1016/j.tet.2017.05.072. Links
-
20Daina, A.; Michielin, O.; Zoete, V. Sci Rep. 2017, 7, 42717. DOI: https://doi.org/10.1038/srep42717. Links
-
21Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. Bioinformatics. 2018, 35, 1067-1069. DOI: https://doi.org/10.1093/bioinformatics/bty707. Links
-
22Qu, R.; Zhang, X.; Zhang, Q.; Yang, X.; Wang, Z.; Wang, L. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 81, 261-269. DOI: https://doi.org/10.1016/j.saa.2011.06.008. Links
-
23Manikandan, D.; Swaminathan, J.; Tagore, S. S.; Gomathi, S.; Sabarinathan, N.; Ramalingam, M.; Balasubramani, K.; Sethuraman, V. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 239, 118484. DOI: https://doi.org/10.1016/j.saa.2020.118484. Links
-
24Foresman, J.; Frish, E. in: Exploring Chemistry with Electronic Structure Methods, Gaussian Inc., Pittsburg, USA, 1996. Links
-
25Sayin, K.; Karakaş, D. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 144, 176-182. DOI: https://doi.org/10.1016/j.saa.2015.02.086. Links
-
26Uluçam, G.; Okan, Ş. E.; Aktaş, Ş.; Yentürk, B. J. Mol. Struct. 2021, 1230, 129941. DOI: https://doi.org/10.1016/j.molstruc.2021.129941. Links
-
27Dennington, R.; Keith, T.; Millam, J. Gauss View, Version 5. Semichem Inc., Shawnee Mission, 2009. Links
-
28Zhen, Y.; Shan, X.; Li, Y.; Lin, Z.; Zhang, L.; Lai, C.; Qin, F. Phytomed. Plus. 2022, 100244. DOI: https://doi.org/10.1016/j.phyplu.2022.100244. Links
-
29Yadav, V.; Krishnan, A.; Baig, M. S.; Majeed, M.; Nayak, M.; Vohora, D. Biophys. Chem. 2022, 285. DOI: https://doi.org/10.1016/j.bpc.2022.106808. Links
-
30Anju, K.; Shoba, G.; Sumita, A.; Balakumaran, M. D.; Vasanthi, R.; Kumaran, R. Spectrochim. Acta, Part A. 2021, 258, 119814. DOI: https://doi.org/10.1016/j.saa.2021.119814. Links
-
31Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L. A. Drug Discovery Today. 2021, 151-164. DOI: https://doi.org/10.1016/j.drudis.2021.09.007. Links
-
32Trott, O.; Olson, A. J. J. Comput. Chem. 2010, 31, 455-461. DOI: https://doi.org/10.1002/jcc.21334. Links
-
33Morris, G. M.; Huey, R.; Lindstrom, W.; Sanner, M. F.; Belew, R. K.; Goodsell, D. S.; Olson, A. J. J. Comput. Chem. 2009, 30, 2785-2791. DOI: https://doi.org/10.1002/jcc.21256. Links
-
34Fatima, A.; Khanum, G.; Sharma, A.; Verma, I.; Arora, H.; Siddiqui, N.; Javed, S. Polycycl. Aromat. Compd. 2023, 43, 1263-1287. DOI: https://doi.org/10.1080/10406638.2022.2026989. Links
-
35Yavuz, S. Ç.; Akkoç, S.; Tüzün, B.; Şahin, O.; Saripinar, E. Synth. Commun. 2021, 51, 2135-2159. DOI: https://doi.org/10.1080/00397911.2021.1922920. Links
-
36Xavier, T.; Kenny, P. T.; Manimaran, D.; Joe, I. H. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 145, 523-530. DOI: https://doi.org/10.1016/j.saa.2015.02.087. Links
-
37Suresh, D.; Amalanathan, M.; Joe, I. H.; Jothy, V. B.; Diao, Y.-P. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 130, 591-603. DOI: https://doi.org/10.1016/j.saa.2014.03.043. Links
-
38Devi, K.S.; Subramani, P.; Parthiban, S.; Sundaraganesan, N. J. Mol. Struct. 2020, 1203, 127403. DOI: https://doi.org/10.1016/j.molstruc.2019.127403. Links
-
39Adwin Jose, P.; Sankarganesh, M.; Dhaveethu Raja, J.; Senthilkumar, G. S.; Nandini Asha, R.; Raja, S. J.; Sheela, C. D. J. Biomol. Struct. Dyn. 2021, 21, 10715-10729. DOI: https://doi.org/10.1080/07391102.2021.1947382. Links
-
40Rodriguez, A. C.; Park, H.-W.; Mao, C.; Beese, L. S. J. Mol. Biol. 2000, 299, 447-462. DOI: https://doi.org/10.1006/jmbi.2000.3728. Links
-
41Williams, R. S.; Green, R.; Glover, J. Nat. Struct. Biol. 2001, 8, 838-842. DOI: https://doi.org/10.1038/nsb1001-838. Links
-
42Rouvinen, J.; Rautiainen, J.; Virtanen, T.; Zeiler, T.; Kauppinen, J.; Taivainen, A.; Mäntyjärvi, R. J. Biol. Chem. 1999, 274, 2337-2343. DOI: https://doi.org/10.1074/jbc.274.4.2337. Links
-
43Okamoto, K.; Ikemori-Kawada, M.; Jestel, A.; von König, K.; Funahashi, Y.; Matsushima, T.; Tsuruoka, A.; Inoue, A.; Matsui, J. ACS Med. Chem. Lett. 2015, 6, 89-94. DOI: https://doi.org/10.1021/ml500394m. Links
-
44Yun, C.-H.; Boggon, T. J.; Li, Y.; Woo, M. S.; Greulich, H.; Meyerson, M.; Eck, M. J. Cancer cell. 2007, 11, 217-227. DOI: https://doi.org/10.1016/j.ccr.2006.12.017. Links
-
45Brough, P. A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J. E.; Cheung, K.-M. J.; Collins, I.; Davies, N. G.; Drysdale, M. J. J. Med. Chem. 2008, 51, 196-218. DOI: https://doi.org/10.1021/jm701018h. Links
-
46Masand, V. H.; Rastija, V. Chemom. Intell. Lab. Syst. 2017, 169, 12-18. DOI: https://doi.org/10.1016/j.chemolab.2017.08.003. Links
-
47DeLano, W. L. http://www.pymol.org, 2002. Links
-
48Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. J. Comput. Chem. 2004, 25, 1605-1612. DOI: https://doi.org/10.1002/jcc.20084. Links
-
49Laskowski, R. A.; Swindells, M. B. J. Chem. Inf. Model. 2011, 51, 2778-2786 DOI: https://doi.org/10.1021/ci200227u. Links
-
50Lipinski, C. A. Drug. Discov. Today. Technol. 2004, 1, 337-41. DOI: https://doi.org/10.1016/j.ddtec.2004.11.007. Links
-
51Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P. W.; Tang, Y. J. Chem. Inf. Model. 2012, 52, 3099-3105. DOI: https://doi.org/10.1021/ci300367a. Links
-
52Saravanan, R.; Seshadri, S.; Gunasekaran, S.; Mendoza-Meroño, R.; García-Granda, S. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 139, 321-328. DOI: https://doi.org/10.1016/j.saa.2014.12.026. Links
-
53Demircioğlu, Z.; Kaştaş, Ç. A.; Büyükgüngör, O. J. Mol. Struct. 2015, 1091, 183-195. DOI: https://doi.org/10.1016/j.molstruc.2015.02.076. Links
-
54Obu, Q. S.; Louis, H.; Odey, J. O.; Eko, I. J.; Abdullahi, S.; Ntui, T. N.; Offiong, O. E. J. Mol. Struct. 2021, 1244, 130880. DOI: https://doi.org/10.1016/j.molstruc.2021.130880. Links
-
55Mumit, M. A.; Pal, T. K.; Alam, M. A.; Islam, M. A.-A.-A.-A.; Paul, S.; Sheikh, M. C. J. Mol. Struct. 2020, 1220, 128715. DOI: https://doi.org/10.1016/j.molstruc.2020.128715. Links
-
56Ouaket, A.; Chraka, A.; Raissouni, I.; El Amrani, M. A.; Berrada, M.; Knouzi, N. J. Mol. Struct. 2022, 1259, 132729. DOI: https://doi.org/10.1016/j.molstruc.2022.132729. Links
-
57Abdou, A.; Omran, O. A.; Nafady, A.; Antipin, I. S. Arabian J. Chem. 2022, 15, 103656. DOI: https://doi.org/10.1016/j.arabjc.2021.103656. Links
-
58Ulucam, G.; Yenturk, B.; Okan, S. E.; Aktas, S. Chem. Pap. 2020, 74, 1881-1889. DOI: https://doi.org/10.1007/s11696-019-01037-9. Links
-
59Almeida, M. O.; Barros, D. A. S.; Araujo, S. C.; Faria, S.; Maltarollo, V. G.; Honorio, K. M. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 184, 169-176. DOI: https://doi.org/10.1016/j.saa.2017.04.070. Links
-
60Boshaala, A.; Said, M. A.; Assirey, E. A.; Alborki, Z. S.; AlObaid, A. A.; Zarrouk, A.; Warad, I. J. Mol. Struct. 2021, 1238, 130461. DOI: https://doi.org/10.1016/j.molstruc.2021.130461. Links
-
61Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K. S.; Ashfaq, M.; Tahir, M. N. J. Mol. Struct. 2022, 1250, 131691. DOI: https://doi.org/10.1016/j.molstruc.2021.131691. Links
-
62Anwer, K. E.; Sayed, G. H.; Ramadan, R. M. J. Mol. Struct. 2022, 1256, 132513. DOI: https://doi.org/10.1016/j.molstruc.2022.132513. Links
-
63Śmiszek-Lindert, W. E.; Chełmecka, E.; Lindert, O.; Dudzińska, A.; Kaczmarczyk-Sedlak, I. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2018, 201, 328-338. DOI: https://doi.org/10.1016/j.saa.2018.05.021. Links
-
64Umar, Y. J. Mol. Struct. 2022, 133230. DOI: https://doi.org/10.1016/j.molstruc.2022.133230. Links
-
65Unsalan, O.; Szolnoki, B.; Toldy, A.; Marosi, G. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 98, 110-115. DOI: https://doi.org/10.1016/j.saa.2012.08.050. Links
-
66Uluçam, G.; Bagcı, U.; Şuekinci Yılmaz, A.; Yentürk, B. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2022, 279, 121429. DOI: https://doi.org/10.1016/j.saa.2022.121429. Links
-
67Mary, Y. S.; Mary, Y. S.; Resmi, K.; Kumar, V. S.; Thomas, R.; Sureshkumar, B. Heliyon. 2019, 5, e02825. DOI: https://doi.org/10.1016/j.heliyon.2019.e02825. Links
-
68Daina, A.; Michielin, O.; Zoete, V. J. Chem. Inf. Model. 2014, 54, 3284-3301. DOI: https://doi.org/10.1021/ci500467k. Links
-
69Daina, A.; Zoete, V. Chem. Med. Chem. 2016, 11, 1117-21. DOI: https://doi.org/10.1002/cmdc.201600182. Links
-
70Walters, W. P.; Murcko, M. A. Adv. Drug Delivery Rev. 2002, 54, 255-71. DOI: https://doi.org/10.1016/S0169-409X(02)00003-0. Links