











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
With the growth of the world population and rapid economic development, as well as the shortage of fossil fuels and global environmental problems that threaten the peaceful existence of mankind and stimulate people's determination to find new energy sources [1,2]. Hydrogen as a renewable and clean energy source has several ideal characteristics, for example, it contains the maximum energy per unit weight, is abundant in the natural environment, and its combustion products do not pollute the environment like sulfur oxidation and aromatic hydrocarbon oxides [3,4]. The U. S. Department of Energy (DOE) system has identified goals for hydrogen storage materials, the average adsorption energy per H2 (average adsorption energy per H2, E ad/H2) in an ideal hydrogen storage material should be between chemisorption and physical adsorption (0.1 - 0.8 eV) to meet the hydrogen storage reversibility of the material [5].
Currently, there are many types of hydrogen storage materials. Carbon-based and chemically active hydrogen storage materials include fullerene, graphene, carbon nanotubes, etc [6,7]. Nano-structured magnesium and magnesium-based hydrides can also be used to store hydrogen through adsorption [8]. In recent years, the semiconductor cluster has become one of the research objects in cluster science and the silicon cluster is the most widely used technology in the semiconductor cluster [9-13].
Silicon is not only rich in content but is also one of the most important materials in the modern industry [11-13]. It has a wide range of applications in various areas of life, such as metallurgy, electronic manufacturing, the military industry, medical, etc [10-12]. But at the same time, the disadvantages of silicon materials are also very obvious. For example, the hollow structure of silicon and the lack of unsaturated bonds in silicon valence electrons with sp 2 hybridization deliver unstable fullerene cages (silicon nanotubes and silicon fullerenes) [10]. However, studies over the years have shown that their structures can be improved by various chemical modifications, such as encapsulating metal atoms in clusters [11] or using H adsorption to enhance the sp 3 bonding between Si atoms to improve stability [12]. Meanwhile, Ryou et al. found that the binding energy of hydrogen molecules to silicon nanotubes is less than 0.1 eV even in stable structures through DFT calculations, and then pure silicon nanotubes are not suitable for hydrogen storage [13]. Kumar et al. found that metal doping enhanced the stability and size selectivity of silicon clusters [14]. Sporea et al. stabilized silicon clusters by encapsulating alkali metal atoms Na, K, and Li inside Si20, which can provide regular shapes for Si20 clusters [15]. Moreover, Ammar et al. investigated the hydrogen storage properties of KSi20 and Ti@KSi20 using the DFT-based B3YLP and M06-2X methods. The adsorption energy values (E̅ ads) per hydrogen molecule satisfy the U.S. DOE targets for hydrogen storage materials [16].
According to our knowledge, NaSi20 should have similar hydrogen storage properties to KSi20. However, the hydrogen storage performance of NaSi20 has not been reported. And more importantly, as previously reported by researchers, Ca, Fe, and Ti metal atoms were often considered and performed well in the previous studies of modified carbon-based and boron-based hydrogen storage materials [17-19]. Meanwhile, Ca is one of the most active atoms in alkaline earth metals, Fe is the transition metal atom with more outer electrons, and Ti is the transition metal atom with fewer outer electrons. To the best of our knowledge, NaSi20 deposited with Ca, Fe, and Ti atoms has not been previously investigated as hydrogen storage materials. In this work, the storage characteristics of H2 on Na-encapsulated Si20 (NaSi20) clusters deposited in Ca, Fe, and Ti were investigated by performing DFT calculations at the theoretical level of B3LYP and M06-2X combined with the 6-311++G(d, p) basis set. The effect of electron properties on NaSi20 fullerenes deposited in Ti, Ca, and Fe was studied. Then the adsorption of n hydrogen molecules (n = 1-6) on Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 is discussed.
Computational methods
To investigate the storage properties of H2 molecules on Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 fullerenes, DFT calculations [20] were performed at the B3LYP/6-311++G(d, p) and M06-2X/6-311++G(d, p) levels of theory for the non-metallic atoms (H, Si) and an effective for the effective pseudopotential basis set LANL2DZ of Na, Ca, Fe and Ti atoms, respectively [20]. Full geometric optimizations were carried out at two levels for free H2 molecules, Si20, NaSi20, Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 substrates as well as nH2-Ca@NaSi20 (n = 1-3), nH2-Fe@NaSi20 (n = 1-4), and nH2-Ti@NaSi20 (n = 1-6) complexes. Because silicon is the most readily available semiconductor in nature, hydrogen can be adsorbed on silicon in both crystalline and amorphous forms and can be desorbed again. Several experimental and theoretical studies have been conducted by Mao WL and Mao HK to investigate whether porous silicon is capable of conducting hydrogen storage [21]. All the calculations were calculated using the Gaussian 09 program [22].
The ionization potential (IP), electron affinity (EA), and chemical hardness (η) can be expressed in equations 1-3, where E(N-1), E(N), and E(N+1) are total energies when the system has N-1, N and N+1 electrons, respectively.
The average binding energy
where Ecluster is the energy of the optimized cluster, ∑E
atom is the sum of the energies of the free atoms of the cluster and n is
total number of the cluster atoms. The binding energies (E
bind) of Ca, Fe, and Ti atoms on NaSi20 substrate clusters are
calculated by equation (5). nH2 molecule
(n = 1-6) adsorption energies (E
ads) and the average adsorption energy of per hydrogen molecule
(
where
The enthalpy difference (ΔH Ɵ) and free energy difference (ΔG Ɵ) of nH2-M@NaSi20 (M = Ca, Fe, and Ti) complexes are calculated by equations (8) and (9), respectively [23].
where,
where
For testing the stability of the wave function of our investigated systems, the keywords of UB3LYP, stable and nosymm are used in the examples for both 4H2/Ti@NaSi20 and 4H2/Fe@NaSi20 complexes and the results show that the wave functions are stable under the perturbations considered. The corresponding figure is given in Supplement 1.
Results and discussion
Geometric structure and electronic properties of Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 clusters
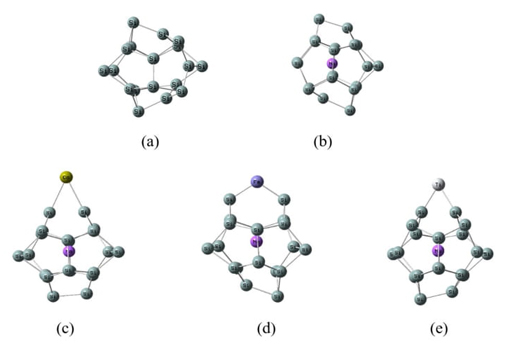
Fig. 1 shows the optimized structures of
Si20 and NaSi20 fullerenes, as well as Ca, Fe, and Ti
external doping NaSi20 fullerenes, named Ca@NaSi20,
Fe@NaSi20, and Ti@NaSi20 clusters. The geometrical and
electronic properties of the investigated clusters are listed in Table 1. As can be seen from Fig. 1(a), the Si20
fullerene cluster is a distorted cage with the shortest Si-Si bond length
(d
Si-Si) of 2.279 Å and dipole moments of 1.198 Debye at the B3LYP
level, and the shortest Si-Si bond length (d
Si-Si) of 2.291 Å and dipole moments of 1.215 Debye at the M06-2X
level. This is similar to the value of Si-Si bond length (d
Si-Si) calculated by Ammar et al., which is 2.335 Å
for Si-Si bond length (d
Si-Si) calculated by B3LYP method and 2.381 Å for Si-Si bond length
(d
Si-Si) calculated by M06-2X method. After a Na atom is embedded in
the Si20 cluster, the structure of the cluster has changed into a
regular shape, as shown in Fig.
1(b). Compared with pure silicon clusters without
internally doped metal atoms, the shape of NaSi20 clusters is more
uniform. Under the B3LYP method, the Si-Si bond length (d
Si-Si) is reduced to 2.777 Å, which is similar to the results
previously reported by Borshch et al. [24]. The HOMO-LUMO gap (E
g) decreased from 2.163 and 3.486 eV to 1.329 and 2.512 eV,
respectively. Chemical hardness (η) decreased by 38.63 % and
27.94 %, respectively, while the average binding energy between atoms
Table 1 Ionization potential (IP, eV), electron affinity (EA, eV), hardness (η, eV), HOMO and LUMO energy gap (E g, eV), binding energy per atom (E b, eV), dipole moment (D, Debye), bond length (d, Å), binding energy (E bind, eV) of Si20, NaSi20, Ca@NaSi20, Fe@NaSi20 and Ti@NaSi20 as well as Mulliken charges (QM, e) of Ca, Fe, Ti atoms in the clusters.
| Si20 | NaSi20 | Ca@NaSi20 | Fe@NaSi20 | Ti@NaSi20 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | M06-2X | |
| IP | 5.780 | 6.581 | 5.255 | 5.922 | 4.761 | 5.485 | 5.372 | 5.656 | 5.209 | 5.391 |
| EA | 3.616 | 3.095 | 3.926 | 3.410 | 3.776 | 3.303 | 3.911 | 3.831 | 3.795 | 3.432 |
| η | 1.082 | 1.743 | 0.664 | 1.256 | 0.493 | 1.091 | 0.730 | 0.913 | 0.707 | 0.979 |
| E g | 2.163 | 3.486 | 1.329 | 2.512 | 0.986 | 2.182 | 1.460 | 1.825 | 1.414 | 1.959 |
| E b | -4.285 | -4.540 | -4.091 | -4.306 | -3.955 | -4.155 | -4.137 | -4.286 | -4.103 | -4.250 |
| D | 1.198 | 1.215 | 0.163 | 0.259 | 11.843 | 11.489 | 2.966 | 1.597 | 8.502 | 9.983 |
| dSi-Si/dM-Si | 2.279 | - | 2.777 | - | 3.013 | - | 2.367 | - | 2.502 | - |
| Ebind | -1.091 | -0.992 | -5.109 | -3.877 | -4.348 | -3.069 | ||||
| QM | 1.266 | 1.042 | -0.184 | -0.123 | 0.398 | 0.466 | ||||
We studied the external doping of Ca, Fe, and Ti atoms at different positions of NaSi20 fullerene. The structure with the most stable energy after optimization is shown in Fig. 1. The results show that Ca, Fe, and Ti are bound to the NaSi20 cluster by bonding with two adjacent silicon atoms, which is the same as the external doping mode of Ti atoms to the KSi20 cluster in the cluster studied by Ammar et al. [14].
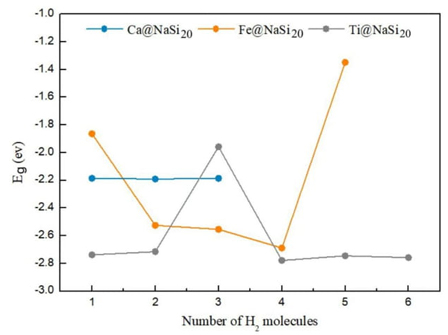
According to Baei's theory, the size of the energy gap (E
g)value is closely related to the activity and stability of chemical
reactions [25]. Under the density
functional B3LYP method, the energy gap (E
g) of the NaSi20 cluster modified by Fe and Ti atoms is
larger than that of the NaSi20 cluster modified by Ca atoms, while
the energy gap (E
g) of the NaSi20 cluster modified by Ca atoms is smaller
than that of the NaSi20 cluster. Similarly, with the B3LYP method,
the chemical hardness (η) of the Fe@NaSi20 cluster
and Ti@NaSi20 cluster increases by 9.93 % and 6.48 %, respectively,
while the chemical hardness (η) of the Ca@NaSi20
cluster decreases by 25.75 %. With the M06-2X method, the Ca@NaSi20,
Fe@NaSi20, and Ti@NaSi20 clusters have smaller energy
gaps than the NaSi20 clusters. The Ca@NaSi20,
Fe@NaSi20, and Ti@NaSi20 clusters also have smaller
η values than the NaSi20 clusters. Under the
M06-2X method, η values of Ca@NaSi20,
Fe@NaSi20 and Ti@NaSi20 are reduced by 22.05 %, 27.31
% and 13.14%, respectively. Since the density functional M06-2X method considers
the weak interaction, it can well describe the weak interaction between metals
and clusters. At the same time, the M06-2X method is effective in calculating
the energy of the reactants, the isomerization process and the reaction energy
barrier. The results showed that the chemical reactivity of
Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 clusters
was better than that of NaSi20 clusters. Compared with
NaSi20 clusters, the average binding energy (
Interaction of H2 on Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20
In this work, hydrogen molecules are stored on optimized Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 clusters to characterize the properties of the selected hydrogen storage materials. Fig. 2 shows the geometry of nH2-Ca@NaSi20, nH2-Fe@NaSi20, and nH2-Ti@NaSi20 clusters and in order to explore the place of adsorption more clearly, the structure diagrams from different viewpoints for 4H2-Fe@NaSi2 and 6H2-Ti@NaSi20 as well as different direction of hydrogen attacking for Ti@NaSi20 and Fe@NaSi20 are given in Fig. 3. As can be seen from Fig. 2, the H-H bonds of all H2 molecules are not broken, and they are all adsorbed by metal atoms in the form of molecules. Parameters such as dipole moment, bond length and Mulliken charge of adsorbed clusters are shown in Tables 2-4, respectively. Equations (6) and (7) were used to calculate the adsorption energy of hydrogen storage materials and the average adsorption energy data of each hydrogen molecule, as shown in Fig. 4(a) and 4(b), respectively. Density functional methods of B3LYP and M06-2X were used to optimize the structure of the H2 molecule, and the results showed that the H-H bond lengths were 0.744 Å and 0.740 Å, respectively.
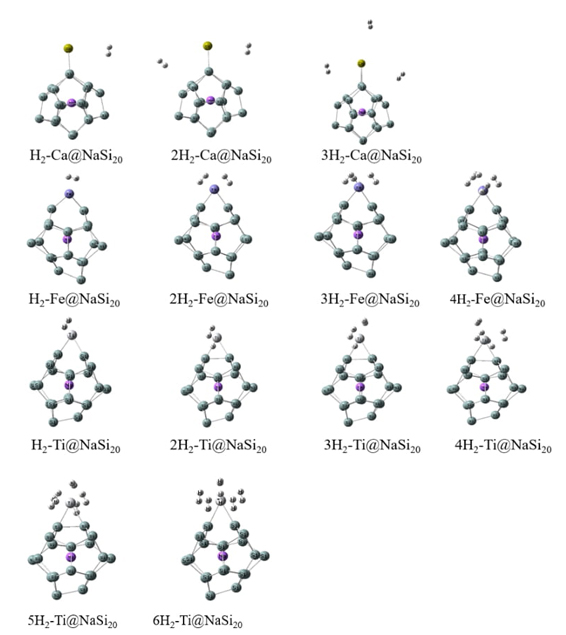
Fig. 2 The optimized structures for nH2-Ca@NaSi20, nH2-Fe@NaSi20 and nH2-Ti@NaSi20 calculated at B3LYP method.
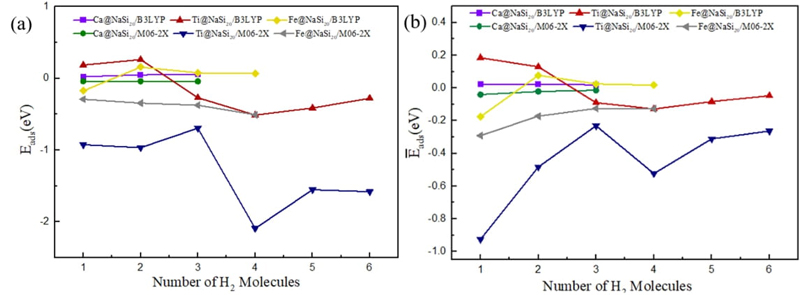
Fig. 3 (a) The adsorption energy (E
ads) and (b) the average adsorption energy per hydrogen
molecule (
Adsorption of H2 on Ca@NaSi20
This section focuses on the interaction between H2 and
Ca@NaSi20 clusters. As can be seen from Table 2, under the B3LYP and M06-2X methods, the interaction
between a single H2 molecule and the Ca@NaSi20 cluster
results in an increase of 0.023 eV and a decrease of 0.040 eV, respectively.
According to Mulliken charge analysis, it can be found that the H2
molecule (
Table 2 The dipole moment (D, Debye), bond length (d, Å), adsorption energy (E ads, eV) and Mulliken charges (Q, e) of Ca@NaSi20.
| D | dCa-Si | dCa-H | dH-H | E ads |
|
QNa | QCa |
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | B3LYP | B3LYP | B3LYP | B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | B3LYP | B3LYP | |
| H2-Ca@NaSi20 | 11.881 | 3.012 | 4.054 | 0.745 | 0.023 | -0.040 | 0.023 | -0.040 | -0.311 | 1.020 | -0.017 |
| 2H2-Ca@NaSi20 | 11.816 | 3.009 | 4.133 | 0.745 | 0.048 | -0.041 | 0.024 | -0.021 | -0.323 | 1.029 | 0.005 |
| 5.552 | 0.745 | -0.019 | |||||||||
| 3H2-Ca@NaSi20 | 11.857 | 3.009 | 4.107 | 0.745 | 0.057 | -0.042 | 0.019 | -0.014 | -0.321 | 1.030 | 0.005 |
| 5.411 | 0.745 | -0.021 | |||||||||
| 6.259 | 0.744 | -0.001 | |||||||||
Adsorption of H2 on Fe@NaSi20
This section concerns the interaction of the H2 molecules with the
Fe@NaSi20 cluster. As can be seen from Table 3, the bond length of the H2 molecule
increases from 0.744 Å to 0.802 Å for a single H2 molecule after
adsorption, increasing by 0.058 Å. After the adsorption of one H2
molecule, the bond length of the Si-Fe atom (dFe-Si) in the
Fe@NaSi20 cluster is 2.320 Å under the B3LYP method, which is
0.047 Å shorter than the Fe@NaSi20 cluster without adsorption of the
H2 molecule. The increasing of hydrogen molecular bond length
(dH-H) and the shortening of Si-Fe atom bond length
(dFe-Si) indicate that the H2 molecule is successfully
adsorbed by the Fe@NaSi20 cluster. In terms of energy range, when the
number of adsorbed H2 molecules increases from 1 to 4, the absolute
value of adsorption energy (E
ads) calculated by the M06-2X method gradually increases from 0.291
eV to 0.508 eV, and the absolute value of average adsorption energy (
Table 3 The dipole moment (D, Debye), bond length (d, Å), adsorption energy (E ads, eV) and Mulliken charges (Q, e) of Fe@NaSi20.
| D | dFe-Si | dFe-H | dH-H | E ads |
|
QNa | QFe |
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | B3LYP | B3LYP | B3LYP | B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | B3LYP | B3LYP | |
| H2-Fe@NaSi20 | 3.359 | 2.320 | 1.771 | 0.802 | -0.175 | -0.291 | -0.175 | -0.291 | -0.408 | -0.298 | 0.119 |
| 2H2-Fe@NaSi20 | 3.397 | 2.260 | 1.629 | 0.851 | 0.156 | -0.345 | 0.078 | -0.172 | -0.346 | -0.434 | 0.296 |
| 1.635 | 0.845 | 0.265 | |||||||||
| 3H2-Fe@NaSi20 | 3.020 | 2.275 | 1.637 | 0.838 | 0.076 | -0.374 | 0.025 | -0.125 | -0.344 | -0.614 | 0.250 |
| 1.635 | 0.842 | 0.230 | |||||||||
| 1.756 | 0.796 | 0.126 | |||||||||
| 4H2-Fe@NaSi20 | 3.083 | 2.289 | 1.636 | 0.843 | 0.067 | -0.508 | 0.017 | -0.127 | -0.368 | -0.630 | 0.257 |
| 1.637 | 0.839 | 0.255 | |||||||||
| 1.769 | 0.793 | 0.005 | |||||||||
| 3.387 | 0.744 | -0.023 | |||||||||
Adsorption of H2 on Ti@NaSi20
This section discusses the interaction of H2 molecules with
Ti@NaSi20 clusters. Table
4 shows the geometric parameters of the optimized
nH2-Ti@NaSi20(n = 6) cluster. Under
the B3LYP method, the bond length (dH-H) of the adsorbed
H2 molecule increases from 0.744 Å to 0.846 Å, and as can be seen
from Fig. 2, there is no break in the
hydrogen bond of all H2 molecules adsorbed by
nH2-Ti@NaSi20(n = 6) system. It
indicates that H2 molecules are adsorbed on the cluster in molecular
form. The Ti-Si atomic bond length (dTi-Si) increases with the number
of H2 adsorbed. When the sixth hydrogen molecule is adsorbed, the
maximum value of the Ti-Si atomic bond length (dTi-Si) is 2.656 Å.
When the seventh molecule is adsorbed by the Ti@NaSi20 cluster, the
Ti-Si bond is broken. That is, the Ti-Si atomic bond length (dTi-Si)
reaches the maximum value when the sixth H2 molecule is adsorbed.
Therefore, the Ti@NaSi20 system can adsorb up to 6 H2
molecules according to the bond length analysis. Under the B3LYP method, the
dipole moment of the H2-Ti@NaSi20 system is 0.250 Debye
smaller than that of Ti@NaSi20, and under the M06-2X method the
dipole moment of the H2-Ti@NaSi20 system is 1.022 Debye
smaller than that of Ti@NaSi20, with the decrease of dipole moment
the stability of the clusters is decreased and the activity is increased.
Meanwhile, Mulliken charge analysis results show that from the analysis of the
charge QTi on the Ti atom, due to the charge transfer and
redistribution around the metal atoms and H2 molecules, compared with
the Ti@NaSi20 cluster, the positive charge of the Ti atom in
H2-Ti@NaSi20 system decreases by 0.045 e, and
QTi gradually decreases as the number of H2 molecules
adsorbs increases, and reaches the minimum value at the adsorption of the sixth
H2 molecule. In addition, Ti atoms have fewer electrons in their
outermost shell and can accept more electrons than Fe atoms, which is one reason
why Ti@NaSi20 can attach more H2 molecules than
Fe@NaSi20 clusters. The absolute values of the adsorption energy
(E
ads) of the first H2 molecule adsorbed by the
Ti@NaSi20 cluster were 0.185 and 0.925 eV, respectively,
calculated by the B3LYP and M06-2X methods. With the number of adsorbed
H2 molecules gradually increasing to 6, the absolute value of
adsorption energy (E
ads) first increases and then decreases, and the absolute value of
average adsorption energy (
Table 4 The dipole moment (D, Debye), bond length (d, Å), adsorption energy (E ads, eV) and Mulliken charges (Q, e) of Ti@NaSi20.
| D | dTi-Si | dTi-H | dH-H | Eads |
|
QNa | QTi |
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | B3LYP | B3LYP | B3LYP | B3LYP | M06-2X | B3LYP | M06-2X | B3LYP | B3LYP | B3LYP | |
| H2-Ti@NaSi20 | 8.252 | 2.465 | 1.957 | 0.791 | 0.185 | -0.925 | 0.185 | -0.925 | -0.344 | 0.353 | -0.155 |
| 2H2-Ti@NaSi20 | 7.850 | 2.452 | 1.870 | 0.824 | 0.261 | -0.966 | 0.130 | -0.483 | -0.347 | 0.297 | -0.131 |
| 1.855 | 0.825 | -0.118 | |||||||||
| 3H2-Ti@NaSi20 | 6.133 | 2.470 | 1.856 | 0.826 | -0.270 | -0.694 | -0.090 | -0.231 | -0.353 | 0.252 | -0.026 |
| 1.855 | 0.840 | -0.007 | |||||||||
| 1.866 | 0.826 | 0.095 | |||||||||
| 4H2-Ti@NaSi20 | 9.268 | 2.546 | 2.020 | 0.776 | -0.512 | -2.091 | -0.128 | -0.523 | -0.366 | 0.279 | -0.036 |
| 2.120 | 0.768 | -0.054 | |||||||||
| 2.028 | 0.775 | 0.094 | |||||||||
| 1.980 | 0.798 | 0.305 | |||||||||
| 5H2-Ti@NaSi20 | 6.426 | 2.535 | 1.912 | 0.814 | -0.413 | -1.552 | -0.083 | -0.311 | -0.404 | 0.220 | -0.021 |
| 1.907 | 0.816 | 0.008 | |||||||||
| 2.014 | 0.793 | 0.101 | |||||||||
| 1.875 | 0.818 | 0.149 | |||||||||
| 1.844 | 0.836 | 0.190 | |||||||||
| 6H2-Ti@NaSi20 | 7.089 | 2.656 | 1.818 | 0.846 | -0.279 | -1.580 | -0.047 | -0.263 | -0.390 | 0.240 | -0.020 |
| 1.916 | 0.811 | 0.041 | |||||||||
| 1.970 | 0.803 | 0.103 | |||||||||
| 1.917 | 0.803 | 0.204 | |||||||||
| 1.995 | 0.796 | 0.191 | |||||||||
| 1.921 | 0.810 | 0.161 | |||||||||
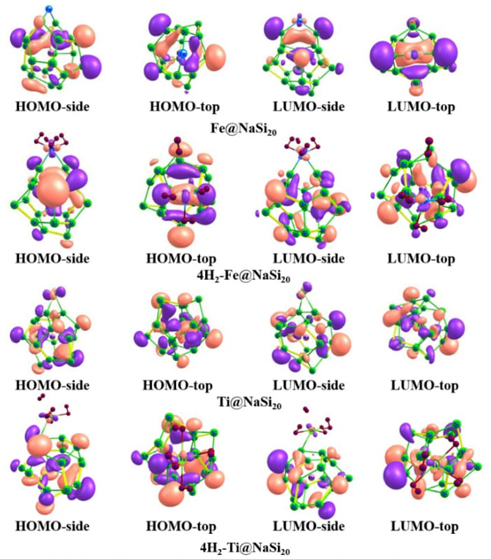
Orbital analysis
The two systems with the best adsorption performance were selected for the wave
function analysis. Fig. 6 shows the
distribution of the highest occupied molecular orbitals (HOMO) and the lowest
unoccupied molecular orbitals (LUMO) in the Fe@NaSi20,
Ti@NaSi20, 4H2-Fe@NaSi20 and
nH2-Ti@NaSi20 (n=3-6) clusters, whose analysis can be used
to guide the design and characterization of novel functional hydrogen storage
fullerenes. The active points are the regions of high local charge density, in which
the red and green colors represent the positive and negative wave functions,
respectively. As can be seen from Fig. 6, in
the Fe@NaSi20 cluster the electron cloud contribution of the Fe atoms in
HOMO-LUMO is almost zero. But after the cluster adsorbed 4 H2 molecules,
the electron cloud around Fe atom in the 4H2-Fe@NaSi20 cluster
increased and HOMO distribution increased, which represents charge transfer between
Fe atoms and H2 molecules. This is consistent with the previous charge
analysis of QFe and
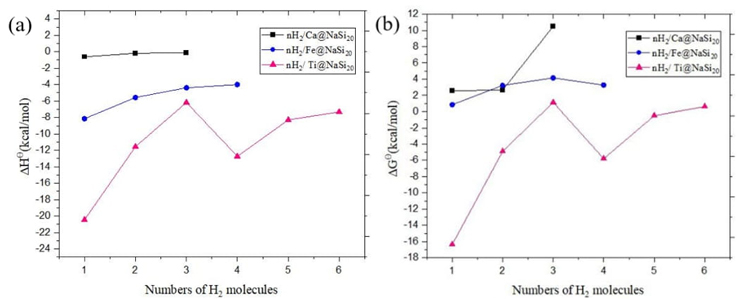
Fig. 6 (a) Enthalpy difference (ΔH Ɵ) and (b) free energy difference (ΔG Ɵ) for nH2/Ca@NaSi20, nH2/Fe@NaSi20 and nH2/Ti@NaSi20 calculated at M06-2X method.
Adsorbed H2 prediction
Theoretically, the Effective Atomic Number Rule (EAN) can be used to calculate the maximum adsorption capacity of hydrogen atoms of hydrogen storage material with surface doped metal atoms [26]. The equation is as follows,
where
Thermodynamic analysis
The enthalpy difference (ΔH Ɵ) can express the strength of the interaction and the free energy difference (ΔG Ɵ) is a measure of the spontaneity of the reaction, which are the key thermodynamic parameters for the interaction between hydrogen and the sorbent materials. ΔH Ɵ and ΔG Ɵ are graphed in Fig. 7 as a function of the number of hydrogen molecules for nH2-Ca@NaSi20, nH2-Fe@NaSi20, and nH2-Ti@NaSi20 complexes. As shown in Fig. 7a, ΔH Ɵ values are negative for all the considered structures, indicating exothermic reactions. For all the complexes of the nH2-Ti@NaSi20 system only the free energy difference of 1H2-Ti@NaSi20, 2H2-Ti@NaSi20, 4H2-Ti@NaSi20, 5H2-Ti@NaSi20 is negative (ΔG Ɵ < 0), which shows that they are spontaneous reactions. In the nH2-Ti@NaSi20 system, when n = 1 the absolute value of ΔHƟ decreases as the number of H2 molecules increases, and the absolute value of ΔH Ɵ is 20.45 kcal/mol. It can be seen from Fig. 7b, in all the complexes, 3H2-Ca@NaSi20 has the largest value of free energy difference (ΔG Ɵ), which is 10.50 kcal/mol. As a negative ΔH Ɵ value indicates stronger stability, and a negative ΔG Ɵ value indicates stronger reversal ability of the reaction, with the increase of the number of hydrogen molecules, the stability decreases, the hydrogen storage capacity gradually reaches saturation, and the desorption ability of H2 molecules increases [27-29].
Conclusions
In this work, the storage characteristics of H2 molecules on NaSi20 fullerenes deposited by Ca, Fe, and Ti were investigated at the theoretical level of the DFT-based B3LYP and M06-2X methods. The results show that the encapsulated Na atom into the Si20 cluster delivers the regular shape to the NaSi20. The deposition of the Ca, Fe and Ti atoms on NaSi20 clusters decreases the ionization potential (IP), HOMO-LUMO energy gap (E g), and hardness (η), and increases the dipole moment (D), which confirms that Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 clusters are less stable and therefore more reactive than NaSi20 clusters.
Additionally, the Ca@NaSi20, Fe@NaSi20, and Ti@NaSi20 clusters are saturated by two, four, and six H2 molecules, respectively. The adsorption energy values (E ads) per hydrogen molecule meet the U. S. DOE target for hydrogen storage materials for nH2-Ti@NaSi20 (n = 2 - 6) and nH2-Fe@NaSi20 (n = 1-4), which implies that NaSi20 fullerenes may be a potentially suitable material for hydrogen storage. The calculated enthalpy differences emphasize that some hydrogen molecules are physisorbed on the studied clusters with an effect greater than one.

