




![Estudios in vitro e in silico de bis-furil-pirrolo[3,4-b]piridin-5-onas en el virus del dengue](/img/es/next.gif)






 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Type 2 diabetes is characterized by a postprandial increase in blood glucose due to the lack of insulin action or secretion, leading to an increased risk of cardiovascular and cerebrovascular diseases, retinopathy, and cancer. According to the World Health Organization (WHO), about 422 million people have diabetes [1]. A therapeutic treatment of diabetes is to reduce blood glucose levels through enzyme inhibitors. The α-amylase and α-glucosidase enzymes are involved in the breakdown of dietary starch and sugars into glucose, where α-amylase catalyzes the hydrolysis of starch and other carbohydrate polymers through the cleavage of 1-4- α-D-glycosidic links generating smaller oligosaccharides while α-glucosidase catalyzes the cleavage of the 1-4- α-D-glycosidic bonds of these oligosaccharides into glucose units [2]. Acarbose is a pseudo-tetrasaccharide that inhibits both enzymes, thus delaying glucose absorption and reducing postprandial glucose levels [3]. However, its gastrointestinal side effects have led to the search for new glucosidase inhibitors from diverse sources, such as natural products and synthetic compounds.
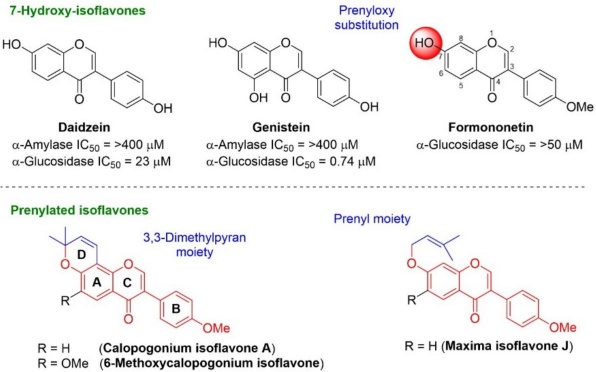
Isoflavones are secondary metabolites of plants, these heterocycles are based on a 3-phenylchroman skeleton, biogenetically derived from a 2-phenylchroman skeleton of flavonoid. They are well-known for their beneficial properties mainly exerting antioxidant [4], antidiabetic [5], anticancer [6], anti-inflammatory [7], anti-ulcer [8], and anti-obesity effects [9]. The main isoflavones sources are soybeans and red clover, with daidzein, genistein, and formononetin as the main compounds. These have attracted attention due to their antidiabetic properties, as they reduce blood glucose levels by acting as α-glucosidase inhibitors [5,10]. Moreover, these natural inhibitors have been employed as lead compounds to obtain new molecules with a higher inhibitory effect on α-glucosidase as well as low adverse effects [7,9,11,12].
Prenylated isoflavones along with pyranoisoflavones are a subclass of natural isoflavones that own at least one prenylated side chain on the flavonoid backbone, exhibiting antimicrobial [13], antidiabetic [14,15], anti-inflammatory [7,16], anticancer [17,18], and neuroprotective properties [12]. Their antidiabetic features are attributed to inhibition of α-glucosidase enzyme [4,14,19,20]. Structure-activity relationship studies of natural isoflavones as α-glucosidase and α-amylase inhibitors suggest that at the C-7 position in the isoflavone core, the presence of a linear or cyclized prenyl moiety seems to favor this effect biological [2,14,20].
The α-glucosidase and α-amylase inhibitory activities shown by the isoflavones daidzein, genistein, and their prenylated derivatives have been reported, but no data are available for prenylated formononetin derivatives (calopogonium isoflavone A, 6-methoxycalopogonium isoflavone and maxima isoflavone J), which is important for the development of dual inhibitors on those carbohydrate-hydrolyzing enzymes (Fig. 1) [11,21]. Considering these facts, the aim of the current study was to synthesize prenylated isoflavones derived-formononetin and test their potential as α-amylase and α-glucosidase inhibitors. Enzymatic and in silico studies were performed to better understand the interaction of the compounds with the active site of both enzymes.
Experimental
Chemistry
All Raw data measurements of melting points were determined by an Electrothermal apparatus and are uncorrected. Nuclear Magnetic Resonance (1H-, 13C-NMR) spectra were recorded on Bruker Avance (600 or 750 MHz) spectrometers. The chemical shifts (() are expressed in ppm relative to the TMS as internal standard and multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), dd (double of doublets), brs (broad singlet), brd (broad doublet), and m (multiplet). A UPLC (Waters) coupled to a quadrupole time-of-flight mass spectrometer (Waters Xevo G2-XS QTOf; electrospray ionization mode ESI-tandem quadrupole) was used for UPLC and mass spectrometer analyses (LC-MS/MS). All the reagents used were acquired from Sigma-Aldrich, and anhydrous solvents were obtained by a distillation process. Analytical TLC was carried out on precoated silica gel plates (Merck 60F254). Silica gel (230-400 mesh) was used for flash chromatography. The synthesis of compounds 1a-c and 2a-c have been previously described [22].
Procedure for the synthesis of 7-hydroxy-isoflavones (2a-c)
A mixture of the corresponding substituted 2,4-dihydroxyacetophenones 1a-c (1.0 mol equiv.) and DMFDMA (2.0 mol equiv.) was poured into at room temperature in a threaded ACE glass pressure tube with a sealed Teflon screw cap. The mixture was heated at 120 °C for 3 h, diluted with CH2Cl2 (30 mL) and the solvent was removed under vacuum. The residue was purified by flash chromatography over silica gel (hexane/EtOAc, 80:20).
7-Hydroxy-3-(4-methoxyphenyl)-4H-chromen-4-one (2a). A crystalline beige solid (1.25 g, 92 %). Rf 0.57 (hexane/EtOAc, 7:3); mp 257-258 ºC. Characterization of 2a has been previously reported in the literature [22].
7-Hydroxy-6-methoxy-3-(4-methoxyphenyl)-4H-chromen-4-one (2b). A beige solid (1.25 g, 80 %). Rf 0.15 (hexane/EtOAc, 7:3); mp 222-224 ºC. Characterization of 2b has been previously reported in the literature [22].
6-Chloro-7-hydroxy-3-(4-methoxyphenyl)-4H-chromen-4-one (2c). A beige solid (2.01 g, 82%). Rf 0.27 (hexane/EtOAc, 7:3); mp 232-233 ºC. Characterization of 2c has been previously reported in the literature [22].
Procedure for the synthesis of pyranoisoflavones (3a-c)
A solution of the corresponding 7-hydroxy-isoflavones (2a-c) (1.0 mol equiv.), 1,1-diethoxy-3-methylbut-2-ene (1.2 mol equiv.), and 3-methylpyridine (0.6 mol equiv.) in xylene (5 mL) was poured into at room temperature in a threaded ACE glass pressure tube with a sealed Teflon screw cap. The mixture was heated at 120 °C for 36 h, diluted with CH2Cl2 (20 mL) and the solvent was removed under vacuum. The residue was purified by flash chromatography over silica gel (hexane/EtOAc, 90:10)
3-(4-Methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3a) [12]. A white solid (0.10 g, 43 %). Rf 0.55 (hexane/EtOAc, 7:3); m.p. 135-137 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.50 (s, 6H, (CH 3)2), 3.84 (s, 3H, OCH3), 5.72 (d, J = 9.75 Hz, 1H, H-9), 6.81 (d, J = 9.75 Hz, 1H, H-10), 6.86 (d, J = 9.0 Hz, 1H, H-6), 6.95-6.99 (m, 2H, H-3’), 7.48-7.51 (m, 2H, H-2’), 7.93 (s, 1H, H-2), 8.06 (d, J = 8.2 Hz, 1H, H-5). 13C-NMR (187.5 MHz, CDCl3) δ: 28.1 ((CH3)2), 55.3 (OCH3), 77.6 (C-8), 109.1 (C-10a), 113.9 (C-3’), 114.9 (C-10), 115.1 (C-6), 118.3 (C-4a), 124.2 (C-1’), 124.6 (C-3), 126.7 (C-5), 130.1 (C-2’), 130.2 (C-9), 151.7 (C-2), 152.3 (C-1a), 157.2 (C-6a), 159.5 (C-4’), 175.8 (C=O). HRMS (ESI) [M-H]- Calculated for: C21H17O4. 333.1127. Found: 333.1120 [M-H]-.
6-Methoxy-3-(4-methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3b) [23]. A white solid (0.074 g, 61 %). Rf 0.33 (hexane/EtOAc, 7:3); m.p. 164-165 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.56 (s, 6H, (CH 3)2), 3.84 (s, 3H, OCH3-C-4’), 3.96 (s, 3H, OCH3-C-6), 5.74 (d, J = 9.75 Hz, 1H, H-9), 6.81 (d, J = 9.75 Hz, 1H, H-10), 6.96-6.99 (m, 2H, H-3’), 7.49-7.52 (m, 2H, H-2’), 7.56 (s, 1H, H-5), 7.95 (s, 1H, H-2). 13C-NMR (187.5 MHz, CDCl3) δ: 27.9 ((CH3)2), 55.3 (OCH3-C-4’), 56.3 (OCH3-C-6), 78.1 (C-8), 105.1 (C-5), 110.1 (C-10a), 113.9 (C-3’), 115.1 (C-10), 117.6 (C-4a), 124.1 (C-3), 124.4 (C-1’), 130.1 (C-2’), 130.3 (C-9), 147.10 (C-6), 147.16 (C-1a), 147.35 (C-6a), 151.5 (C-2), 159.4 (C-4’), 175.5 (C=O). HRMS (ESI) [M-H]- Calculated for: C22H19O5. 363.1232. Found: 363.1225 [M-H]-.
6-Chloro-3-(4-methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3c). A white solid (0.09 g, 53 %). Rf 0.50 (hexane/EtOAc, 7:3); m.p. 150-151 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.55 (s, 6H, (CH 3)2), 3.83 (s, 3H, OCH3), 5.78 (d, J = 9.75 Hz, 1H, H-9), 6.80 (d, J = 9.75 Hz, 1H, H-10), 6.95-6.99 (m, 2H, H-3’), 7.45-7.50 (m, 2H, H-2’), 7.93 (s, 1H, H-2), 8.12 (s, 1H, H-5). 13C-NMR (187.5 MHz, CDCl3) δ: 28.0 ((CH3)2), 55.3 (OCH3), 79.0 (C-8), 110.7 (C-10a), 113.9 (C-3’), 114.7 (C-10), 118.4 (C-6), 120.4 (C-4a), 123.8 (C-1’), 124.6 (C-3), 126.0 (C-5), 130.0 (C-2’), 130.8 (C-9), 150.5 (C-1a), 151.8 (C-2), 152.6 (C-6a), 159.6 (C-4’), 174.9 (C=O). HRMS (ESI) [M-H]- Calculated for: C21H16ClO4. 367.0737. Found: 367.1691.
Procedure for the synthesis of chromenes (4a-c)
Following the method of preparation for 3a, a mixture of the corresponding 2,4-dihydroxyacetophenone (1a-c) (1.0 mol equiv.), 1,1-diethoxy-3-methylbut-2-ene (1.2 mol equiv.), and 3-methylpyridine (0.6 mol equiv.) in xylene (3 mL) was heated at 120 °C for 12 h. The residue was purified by flash chromatography over silica gel (hexane/EtOAc, 98:2).
1-(5-hydroxy-2,2-dimethyl-2H-chromen-6-yl)-2-(4-methoxyphenyl)ethan-1-one (4a) [24]. Yellow crystals (0.178 g, 71 %). Rf 0.63 (hexane/EtOAc, 8:2); m.p. 115-116 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.37 (s, 6H, (CH 3)2), 3.71 (s, 3H, OCH3), 4.06 (s, 2H, CH 2-H-2’), 5.49 (d, J = 10.2 Hz, 1H, H-3), 6.26 (d, J = 9.0 Hz, 1H, H-8), 6.62 (d, J = 10.2 Hz, 1H, H-4), 6.77-6.83 (m, 2H, H-3’’), 7.07-7.13 (m, 2H, H-2’’), 7.56 (d, J= 9.0 Hz, 1H, H-7), 12.88 (s, 1H, OH). 13C-NMR (187.5 MHz, CDCl3) δ: 28.3 ((CH3)2), 43.8 (CH2-C-2’), 55.2 (OCH3), 77.7 (C-2’), 108.3 (C-8), 109.3 (C-4a), 112.9 (C-6), 114.1 (C-3’’), 115.6 (C-4), 126.3 (C-1’’), 128.1 (C-3), 130.3 (C-2’’), 131.3 (C-7), 158.5 (C-4’’), 159.7 (C-8a), 160.0 (C-5), 202.4 (C=O). HRMS (ESI) [M-H]- Calculated for: C20H19O4. 323.1289. Found: 323.1295.
1-(5-hydroxy-8-methoxy-2,2-dimethyl-2H-chromen-6-yl)-2-(4-methoxyphenyl)ethan-1-one (4b) [25]. Yellow crystals (0.15 g, 65 %). Rf 0.44 (hexane/EtOAc, 8:2); m.p. 177-178 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.50 (s, 6H, (CH 3)2), 3.79 (s, 3H, OCH3-C-4’), 3.81 (s, 3H, OCH3-C-8), 4.13 (s, 2H, CH 2-H-2’), 5.59 (d, J = 9.75 Hz, 1H, H-3), 6.70 (d, J = 9.75 Hz, 1H, H-4), 6.86-6.90 (m, 2H, H-3’’), 7.15 (s, 1H, H-7), 7.17-7.20 (m, 2H, H-2’’), 12.76 (s, 1H, OH). 13C-NMR (187.5 MHz, CDCl3) δ: 28.2 ((CH3)2), 44.3 (CH2-C-2’), 55.2 (OCH3-C-4’), 57.0 (OCH3-C-8), 78.2 (C-2), 110.4 (C-4a), 111.0 (C-6), 112.9 (C-7), 114.2 (C-3’’), 115.9 (C-4), 126.4 (C-1’’), 128.3 (C-3), 130.2 (C-2’’), 141.1 (C-8), 150.3 (C-8a), 155.5 (C-5), 158.6 (C-4’’), 201.9 (C=O). HRMS (ESI) [M-H]- Calculated for: C21H21O5. 353.1394. Found: 353.1378.
1-(8-chloro-5-hydroxy-2,2-dimethyl-2H-chromen-6-yl)-2-(4-methoxyphenyl)ethan-1-one (4c). A yellow solid (0.143 g, 58 %). Rf 0.66 (hexane/EtOAc, 8:2); m.p. 140-142 °C. 1H-NMR (750 MHz, CDCl3) δ: 1.49 (s, 6H, (CH 3)2), 3.79 (s, 3H, OCH3-C-4’), 4.12 (s, 2H, CH 2-H-2’), 5.61 (d, J = 9.75 Hz, 1H, H-3), 6.68 (d, J = 9.75 Hz, 1H, H-4), 6.86-6.90 (m, 2H, H-3’’), 7.14-7.19 (m, 2H, H-2’’), 7.70 (s, 1H, H-7), 12.75 (s, 1H, OH). 13C-NMR (187.5 MHz, CDCl3) δ: 28.3 ((CH3)2), 43.8 (CH2-C-2’), 55.2 (OCH3-C-4’), 79.1 (C-2), 110.8 (C-4a), 112.3 (C-8), 113.1 (C-6), 114.2 (C-3’’), 115.5 (C-4), 125.7 (C-1’’), 128.6 (C-3), 130.3 (C-2’’), 130.5 (C-7), 155.0 (C-8a), 158.3 (C-5), 158.7 (C-4’’), 202.0 (C=O). HRMS (ESI) [M-H]- Calculated for: C20H18ClO4. 357.0899. Found: 357.0861.
Procedure for the synthesis of pyranoisoflavones (3a-c). Following the method for preparation for 2a, a mixture of the corresponding chromene (4a-c) (1.0 mol equiv.) and DMFDMA (2.0 mol equiv.) was heated at 120 °C for 12 h. The residue was purified by flash chromatography over silica gel (hexane/EtOAc, 90:10).
3-(4-Methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3a). A white solid (0.05 g, 50 %), Rf 0.55 (hexane/EtOAc, 7:3); m.p. 135-137 °C.
6-Methoxy-3-(4-methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3b). A white solid (0.046 g, 47 %), Rf 0.33 (hexane/EtOAc, 7:3); m.p. 164-165 °C.
6-Chloro-3-(4-methoxyphenyl)-8,8-dimethyl-4H,8H-pyrano[2,3-f]chromen-4-one (3c). A white solid (0.055 g, 55 %), Rf 0.50 (hexane/EtOAc, 7:3); m.p. 150-151 °C.
General Method for the preparation of 7-O-prenyl-isoflavones (5a-c)
At room temperature (rt), 3,3-dimethylallyl bromide (1.5 mol equiv.) was added dropwise to a solution of 7-hydroxy-isoflavone 2a-c (1.0 mol equiv.), and K2CO3 (2.0 mol equiv.) in dried acetone (20 mL), then stirred for 15 minutes. The reaction mixture was refluxed at 60 °C for 3 h, filtered, and the solvent removed under vacuum. The crude residue was purified by flash chromatography over silica gel (hexane/EtOAc, 80:20)
3-(4-Methoxyphenyl)-7-((3-methylbut-2-en-1-yl)oxy)-4H-chromen-4-one (5a) [7]. A white solid (0.434 g, 87 %). Rf 0.57 (hexane/EtOAc, 7:3), mp 131-132 °C. 1H NMR (750 MHz, DMSO-d 6 ): δ 1.74 (s, 3H, H-4’’), 1.76 (s, 3H, H-5’’), 3.79 (s, 3H, CH 3O), 4.68 (sbr, 2H, CH 2-H-1’’), 5.47 (tbr, 1H, CH-H2’’), 6.90-7.10 (m, 3H, H-3’, H-6), 7.06 (dbr, J = 6.7 Hz, 1H, H-8), 7.43-7.60 (m, 2H, H-2’), 8.01 (dbr, J = 6.7 Hz 1H, H-5), 8.41 (sbr, 1H, H-2). 13C NMR (187.5 MHz, DMSO-d 6 ): δ 18.5 (C-5’’), 25.4 (C-4’’), 55.1 (OCH3), 65.3 (OCH2-C1’’), 101.2 (C-8), 113.6 (C-3’), 115.2 (C-6), 117.4 (C-4a), 118.9 (C-2’’), 123.3 (C-3), 124.0 (C-1’), 126.8 (C-5), 130.0 (C-2’), 138.3 (C-3’’), 153.4 (C-2), 157.3 (C-8a), 159.0 (C-4’), 162.8 (C-7), 174.6 (CO-4). HRMS (ESI) [M-H]- Calculated for: C21H19O4: 335.1283. Found: 335.1302 [M-H]-.
6-Methoxy-3-(4-methoxyphenyl)-7-((3-methylbut-2-en-1-yl)oxy)-4H-chromen-4-one (5b). A white solid (0.177 g, 72 %). Rf 0.36 (hexane/EtOAc, 7:3), mp 156-157 °C. 1H NMR (600 MHz, CDCl3): δ 1.71 (s, 3H, H-4’’), 1.74 (s, 3H, H-5’’), 3.76 (s, 3H, CH 3O-H-4’), 3.89 (s, 3H, CH 3O-H-6), 4.61 (d, J = 6.6 Hz, 2H, CH 2-H-1’’), 5.47 (t, J = 6.6 Hz, 1H, CH-H2’’), 6.79 (s, 1H, H-8), 6.86-6.92 (m, 2H, H-3’), 7.39-7.46 (m, 2H, H-2’), 7.54 (s, 1H, H-5), 7.85 (s, 1H, H-2). 13C NMR (150 MHz, CDCl3): δ 18.3 (C-5’’), 25.8 (C-4’’), 55.2 (OCH3-C4’), 56.2 (OCH3-C6), 66.2 (OCH2-C1’’), 100.5 (C-8), 104.9 (C-5), 113.9 (C-3’), 117.7 (C-4a), 118.5 (C-2’’), 124.3 (C-1’), 124.5 (C-3), 130.1 (C-2’), 139.0 (C-3’’), 148.0 (C-6), 151.7 (C-2), 152.1 (C-8a), 153.6 (C-4’), 159.5 (C-7), 175.5 (CO-4). HRMS (ESI) [M-H]- Calculated for: C22H21O5: 365.1389. Found: 365.1386.
6-Chloro-3-(4-methoxyphenyl)-7-((3-methylbut-2-en-1-yl)oxy)-4H-chromen-4-one (5c). A white solid (0.206 g, 84 %). Rf 0.52 (hexane/EtOAc, 7:3), mp 166-167 °C. 1H NMR (600 MHz, DMSO-d 6 ): δ 1.76 (s, 3H, H-4’’), 1.79 (s, 3H, H-5’’), 3.79 (s, 3H, CH 3O), 4.78 (d, J = 6.6 Hz, 2H, CH 2-H-1’’), 5.50 (t, J = 6.6 Hz, 1H, CH-H2’’), 6.97-7.03 (m, 2H, H-3’), 7.43 (s, 1H, H-8), 7.50-7.57 (m, 2H, H-2’), 8.05 (s, 1H, H-5), 8.49 (s, 1H, H-2). 13C NMR (150 MHz, DMSO-d 6 ): δ 18.2 (C-5’’), 25.5 (C-4’’), 55.1 (OCH3), 66.6 (OCH2-C1’’), 102.4 (C-8), 113.6 (C-3’), 117.6 (C-4a), 118.4 (C-2’’), 120.3 (C-3), 123.3 (C-1’), 123.7 (C-6), 125.7 (C-5), 130.0 (C-2’), 139.1 (C-3’’), 153.8 (C-2), 155.8 (C-8a), 157.7 (C-7), 159.1 (C-4’), 173.7 (CO-4). HRMS (ESI) [M-H]- Calculated for: C21H18ClO4: 369.0894. Found: 369.0909.
Inhibition of α-glucosidase
A reaction was prepared by mixing 20 µL α-glucosidase solution (0.5 unit/mL), 120 µL of 0.1 M phosphate buffer (pH 6.9), and 10 µL of the samples at concentrations from 400 µM to 4.0 µM. The solution was incubated in a 96-well microplate at 37 °C for 15 min. Then the enzymatic reaction was initiated by adding 20 µL of 5 mM p-NPG solution in 0.1 M phosphate buffer (pH 6.9), followed by incubation at 37 °C for 15 min. The reaction was stopped by adding 80 µL of 0.2 M Na2CO3 and absorbance was read at 405 nm [22]. Acarbose was used as reference control. IC50 is the concentration that gives 50 % α-glucosidase inhibition.
Inhibition of α-amylase
The reaction mixture consisting of 50 µL of 0.1 M phosphate buffer (pH 6.8), 10 µL of α-amylase solution (5.0 unit/mL), and 20 µL of the sample at various concentrations (from 100 µM to 5.0 µM) was placed in a 96-well plate and pre-incubated at 37 °C for 15 min, and 20 µL of 1 % soluble starch (0.1 M phosphate buffer, pH 6.8) was then added as a substrate and incubated at 37 °C for 45 min. Finally, 100 µL of 3,5-dinitrosalicylic acid (DNS) was added and heated at 100 °C for 20 min, and absorbance was then read at 540 nm [26]. Acarbose was used as reference control. IC50 is the concentration that gives 50 % α-glucosidase inhibition.
Kinetic study
Kinetic studies were carried out with α-glucosidase and α-amylase using a methodology such as that describe in the inhibitory activity assays. The prenylated isoflavone was evaluated at two concentrations according to their IC50. Various concentrations of substrates were used for each of the enzymes in the range of 0.5-5.0 mM for p-NPG in α-glucosidase and 0.1-1.0 % for α-amylase. The type of inhibition for this compound was determined by utilizing double reciprocal plots. Inhibition constants (KI) were calculated from substrate versus reaction rate curves using nonlinear regression of the enzyme inhibition kinetic function [22].
Docking studies
The molecular docking studies were carried out in the AutoDock 4 program [27] using the crystallized proteins of isomaltase from Saccharomyces cerevisiae (PDB: 3A4A) and human pancreatic α-amylase (PDB: 1B2Y) in complex with the inhibitor acarbose. In these proteins, water molecules were removed, hydrogen atoms were added to the polar atoms (considering pH at 7.4), and Kollman charges were assigned with AutoDock Tools 1.5.6. The 3D structures of acarbose (5) and isoflavone derivative 5c were sketched in two dimensions (2D) with ChemSketch and then converted to 3D in a mol2 format using the Open Babel GUI program [28]. The ligands were optimized with PM6 on Gaussian 98 software to obtain the lowest energy conformation. All the possible rotatable bonds, torsion angles, atomic partial charges, and non-polar hydrogens were determined for each ligand. In AutoDockTools, the grid dimensions for α-glucosidase were 78 × 60 × 78 Å3 with points separated by 0.375 Å and centered at X = 26.313, Y = -3.544, and Z = 26.146. The grid dimensions for α-amylase were 90 × 70 × 66 Å3 with points separated by 0.375 Å and centered at X = 16.758, Y = 8.692, and Z = 49.959. The hybrid Lamarckian genetic algorithm was applied for minimization and utilized default parameters. A total of one hundred docking runs were conducted to determine the conformation with the lowest binding energy (kcal/mol), which was adopted for all further simulations. AutoDockTools was used to prepare the script and files as well as to visualize the docking results, and these were edited with Discovery Studio Visualizer [29].
Results and discussion
Chemistry
Pyranoisoflavones are generally obtained using hydroxyketones as building blocks through two approaches: i) synthesis of hydroxylated isoflavones and formation of the 3,3-dimethyl pyran ring; and ii) synthesis of benzopyran moiety followed by the assembling of the isoflavone core [7]. In order to evaluate the pyran moieties within the isoflavone core, both strategies were applied and the synthesis of prenylated isoflavones is described in scheme 1.
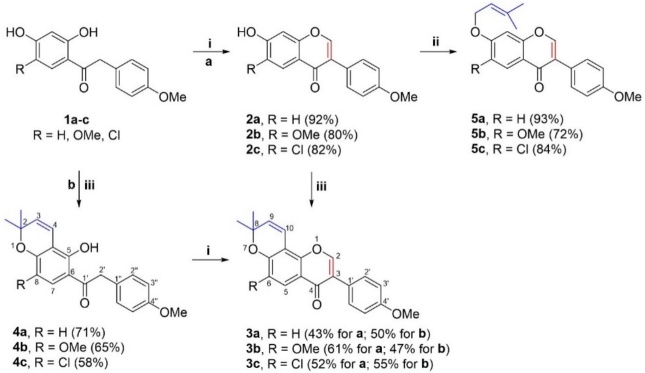
Scheme 1 Synthesis of prenylated isoflavone derivatives. Reagents and conditions: (i) DMFDMA, 120 °C, 3-12 h: (ii) 3,3-dimethylallyl bromide, acetone, K2CO3, 60 °C, 3 h; (iii) 1,1-diethoxybut-2-ene, 3-methylpicoline, xylene, 120 °C, 24-36 h.
In the first case, 7-hydroxyisoflavones derivatives 2a-c were prepared in good yields by treatment of 2,4-dihydroxyacetophenones 1a-c with DMFDMA under thermal conditions [22]. Then, the cyclization of 7-hydroxyisoflavones 2a-c with 1,1-diethoxy-3-methylbut-2-ene in presence of 3-methylpicoline provided the pyranoisoflavones calopogonium isoflavone A (3a), 6-methoxycalopogonium isoflavone (3b), and 6-chloro-pyranoisoflavone (3c) in moderate to good yields. On the other hand, the chromene derivatives 4a-c were obtained by cyclization of 2,4-dihydroxyketones 1a-c with 1,1-diethoxy-3-methylbut-2-ene in presence of 3-methylpicoline, which were cyclized with DMFDMA giving the corresponding pyranoisoflavones 3a-c. Finally, the O-alkylation reaction of isoflavones 2a-c with 3,3-dimethylallyl bromide generated the series of 7-prenyloxy-isoflavones 5a-c in good yields [7].
Pyranoisoflavones calopogonium isoflavone A (3a), 6-methoxycalopogonium isoflavone (3b), and 7-prenyloxyisoflavone maxima isoflavone J (5a) have been isolated from plants of the genus Millettia and Placolobium (Fabaceae) [23,30-33]. Recently, compound 4b has been isolated from P. vietnamense and has been given the name Placovinone D [25]. The elucidation of the natural and synthetic prenylated isoflavones was confirmed by NMR and HRMS techniques, and the spectroscopic data of 3a-b, 4b, and 5a were in accordance with the published literature [7,12].
In vitro α-glucosidase inhibition
The inhibitory activity of α-glucosidase was assessed for all the synthesized compounds. The obtained IC50 values were compared to the corresponding value of acarbose (6), a well-known drug that inhibits α-glucosidase and α-amylase enzymes. As summarized in Table 1, most of the compounds display significant α-glucosidase inhibition with IC50 values in the range of 17.69 to 391.46 µM in comparison to 6 with IC50 of 527.5 µM. An exception was compound 3b, which showed IC50 >400 µM. 7-hydroxyisoflavones 2a-c showed good inhibitory activity whereas compound 2c (IC50 = 91.99 ± 0.21 µM) produced a better effect of almost 6-fold higher than 6. The addition of a cyclized prenyl moiety at C-7 - C-8 (E-ring) of the 7-hydroxyisoflavone backbone led to a significant loss of inhibitory effect with IC50 values of 208.0 ± 0.45 and 260.1 ± 0.52 µM (compounds 3a and 3c). Interestingly, chromene benzyl derivatives 4a-c (without C-ring) displayed greater inhibition that 3a-c, where 4c exhibited 9-fold higher than 6. In order to explore that linear prenyl moiety favors inhibition, derivatives 5a-c were evaluated. These latter compounds exhibited better inhibitory effect than pyranoisoflavones (3a-c), being 5c as the most potent derivative (IC50 = 17.7 ± 0.02 µM), with almost 30-fold greater inhibition of α-glucosidase than 6.
Table 1 α-Glucosidase and α-amylase inhibition by the test compounds.
| Compound | α-Glucosidase inhibition | α-Amylase inhibition | ||
| % (400 µM) | IC50 (µM) | % (100 µM) | IC50 (µM) | |
| 2a | 94.21 ± 0.91 | 95.78 ± 0.04F | 63.53 ± 2.55 | 70.68 ± 1.2D |
| 2b | 92.0 ± 0.15 | 111.4 ± 0.23F | 30.69 ± 2.65 | >100 |
| 2c | 99.65 ± 0.26 | 91.99 ± 0.21FG | 81.93 ± 1.37 | 56.87 ± 0.7E |
| 3a | 59.34 ± 1.01 | 208.0 ± 0.45D | 92.56 ± 3.70 | 42.23 ± 0.9F |
| 3b | 32.59 ± 0.52 | >400 | 6.45 ± 1.25 | -a |
| 3c | 93.30 ± 0.61 | 260.1 ± 0.52C | 99.05 ± 0.51 | 34.96 ± 1.18F |
| 4a | 99.38 ± 0.05 | 78.01 ± 0.21H | 99.44 ± 0.17 | 123.20 ± 0.03C |
| 4b | 94.83 ± 0.15 | 147.65 ± 0.15B | 8.69 ± 0.04 | - |
| 4c | 99.64 ± 0.03 | 57.30 ± 0.05I | 70.61 ± 0.22 | 143.10 ± 0.07G |
| 5a | 97.00 ± 1.65 | 60.56 ± 0.14GH | 99.33 ± 0.93 | 85.09 ± 1.7B |
| 5b | 51.74 ± 1.67 | 391.46 ± 0.18E | 6.81 ± 2.43 | - |
| 5c | 99.15 ± 0.30 | 17.69 ± 0.02H | 98.33 ± 0.07 | 21.2 ± 0.1A |
| Acarbose | 45.65 ± 1.0 | 527.5 ± 0.6A | 98.01 ± 0.25 | 20.18 ± 1.48G |
Data represent the mean + standard deviation (n = 4). Means in a column not sharing the same letter are significantly different at p ˂ 0.5 probability according to Tukey tests; a Not active (less than 30% inhibition at 400 µM).
Regarding the inhibitory effect of the cyclized prenyl moiety (D-ring) on the 7-hydroxyisoflavone core, the analysis of the structure-activity relationship revealed that the dimethylpyran group in the pyranoisoflavone (3a-c) presented a weak inhibitory behavior. In contrast, chromene benzyl derivatives 4a-c, compounds without the pyran ring (C-ring) exhibited greater inhibition, which indicates that the hydroxy group contributed to a higher inhibitory effect. These data suggest that the incorporation of a cyclized prenyl moiety to the isoflavone core significantly decreased the α-glucosidase inhibition, while a prenyloxy moiety at C-7 enhanced inhibitory activity. Furthermore, the presence of the chlorine atom at C-6 increased the inhibitory effects, while that the methoxy group decreased the activity. These results are in accordance with Jo et al. who reported that the α-glucosidase inhibitory activity was stronger in isoflavones with a linear prenyl group than cyclized ones [2,14].
In vitro α-amylase inhibition
All the synthesized compounds were assessed for their inhibition of α-amylase. The IC50 values obtained were compared to the corresponding value of acarbose (6) (Table 1). In general, almost all compounds showed good to weak α-amylase activity with IC50 values in the range of 21.2 to 143.1 µM compared to 6 (IC50 = 20.18 ± 1.48 µM). Derivatives 2b, 3b, 4b, and 5b did not show activity at 100 µM. Initially, 7-hydroxy-isoflavones 2a and 2c showed moderate to weak inhibitory effects. The incorporation of the 3,3-dimethylpyrano group (D-ring) to the isoflavone core displayed almost two-fold greater effect than their parent compounds 2a and 2c. Contrarily, derivatives 4a and 4c were the less effective compounds, presenting IC50 values of 123 2 ± 0.03 and 143.1 ± 0.07 µM, respectively. In addition, the presence of the prenyloxy group at C-7 (compound 5a) resulted in a lower inhibitory effect compared to 2a. Interestingly, compound 5c (IC50 = 21.2 ± 0.1 µM) significantly increased the inhibitory effect concerning 2c. Therefore, the presence of a chlorine atom at C-6 of the isoflavone core significantly enhanced the inhibition, while with the methoxy group did not show any inhibitory activity. These observations suggest that by introducing a linear or cyclized prenyl moiety into the isoflavone core, the inhibitory effect of α-amylase was enhanced.
In summary, the incorporation of a cyclized prenyl moiety (3a and 3c) at the isoflavone scaffold exhibited moderate inhibition of α-glucosidase and α-amylase, while that compounds 4a and 4c (without C-ring) displayed higher inhibition of α-glucosidase and lowest inhibition of α-amylase. On the other hand, a greater inhibition of α-glucosidase with a moderate inhibition of α-amylase were found by addition of a prenyloxy moiety (5a and 5c).
Enzymatic kinetic study
In order to explore the mechanism of interaction of 5c with α-glucosidase and α-amylase, the type of inhibition was evaluated by analyzing Lineweaver-Burk (double reciprocal) plots. The X-axis values represent the reciprocal for the α-glucosidase substrate, p-nitrophenyl-α-D-glucopyranoside (p-NPG), thus being 1/(p-NPG), while for the α-amylase substrate, starch. Thus being 1/(starch). The Y-axis value are the reciprocal of the reaction velocity (Vo), thus being 1/Vo. Given that the plots intersect the Y-axis, the inhibition for α-glucosidase exerted by this compound is carried out in competitive mode (Fig. 2(a) and 2(b)). The Ki value of 5c is 28.5 µM. The Ki value for this compound is less than Km, indicating that it has a higher affinity for the enzyme than the substrate used in the assay.
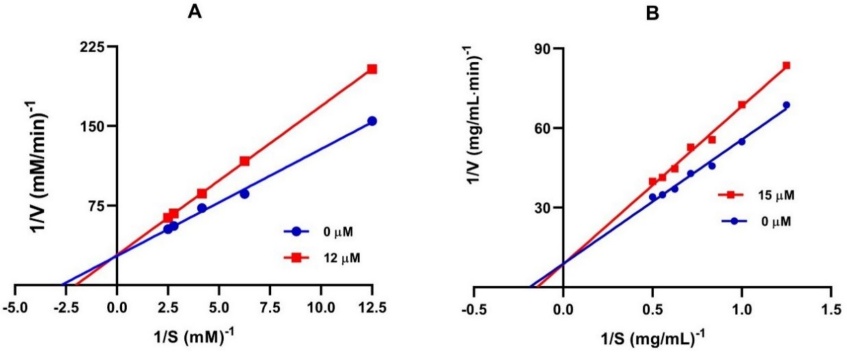
The α-amylase plot made it possible to determine that 5c is a competitive type of inhibitor. Its Ki value (15.2 µM) indicates that has greater affinity for the enzyme.
Molecular docking analysis
To explore the binding interactions of the most active compound, molecular docking studies of the isoflavone derivative 5c and the enzymes isomaltase (α-glucosidase from S. cerevisiae) and the human α-amylase were carried out. The results are illustrated in 2D and 3D (Fig. 3(a) and 3(b)), revealing that 5c recognized some of the key amino acid residues in the catalytic pocket, such Arg213, Asp215, Glu277, His351, Asp352, and Arg442 [22,34]. Regarding the binding energy of the compound 5c with the enzymes, has better binding energy values (ΔG) than the reference drug (6) (Table 2). The docking studies with α-glucosidase reveal that isoflavone system is involved in hydrophobic interactions of various types: π-π-T-shaped (Tyr158), π-anion (Asp352), π-cation (Arg442), and π-sigma (Tyr172), as well as hydrogen bond interactions with Asp215 and Asp352. The prenyloxy moiety at C-7 of the isoflavone core shows a hydrophobic interaction with Phe314 and a hydrophilic interaction with Arg315. According to these results, 5c, interacts with at least two of the acid residues of the catalytic triad, confirming the competitive inhibition of this compound.
Table 2 Docking results of 5c and acarbose (6) at the active site of isomaltase and α-amylase.
| Compound | Binding energy ΔG (kcal/mol) | Interacting residues | Polar interactions | Hydrophobic interactions |
| Isomaltase | ||||
| 6 | -7.78 | Asp69, Tyr72, His112, Tyr158, Phe159, Phe178, Arg213, Asp215, Val216, Glu277, Gln279, His280, Phe303, Asp307, Arg315, Tyr316, His351, Asp352, Gln353, Glu411, Arg442, Arg446 |
C-H…..O (Asp69)
O-H…..O (Aps215) O…..H-N (Gln279) O-H…..O (Asp307) O…..H-N (His351) O-H…..O (Glu411) O…..H-N (Arg446) |
- |
| 5c | 8.62 | Tyr72, Lys156, Ser157, Tyr158, Phe159, Phe178, Arg213, Asp215, Val216, Glu277, Gln279, His280, Phe303, Leu313, Phe314, Arg315, Tyr316, His351, Asp352, Gln353, Glu411, Asn415, Arg442 | C-H…..O (Asp215)
O…..H-N (Arg315) C-H…..N (His351) C-H…..O (Asp352) |
π-sigma-Tyr172
Alkyl-Lys156 π-π T-shaped-Tyr158 π-alkyl-Phe314, Arg315, Tyr316 π-anion-Asp352 π-cation-Arg442 |
| α-Amylase | ||||
| 6 | -2.92 | Asp197, Glu233, Asp300, His305 | O-H…...O (Asp197)
O-H…...O (Glu233) C-H…...O (Glu233) O-H…...O (Asp300) C-H…...O (Asp300) O…..H-N (His305) |
- |
| 5c | -8.25 | Trp58, Trp59, Tyr62, Gln63, Gly104, Val107, Thr163, Leu165, Arg195, Asp197, Glu233, His299, Asp300, His305 | - | π-alkyl-Trp59
π-π stacked-Trp59, Tyr62 π-sigma-Tyr62 Alkyl-Val107, Leu165 |
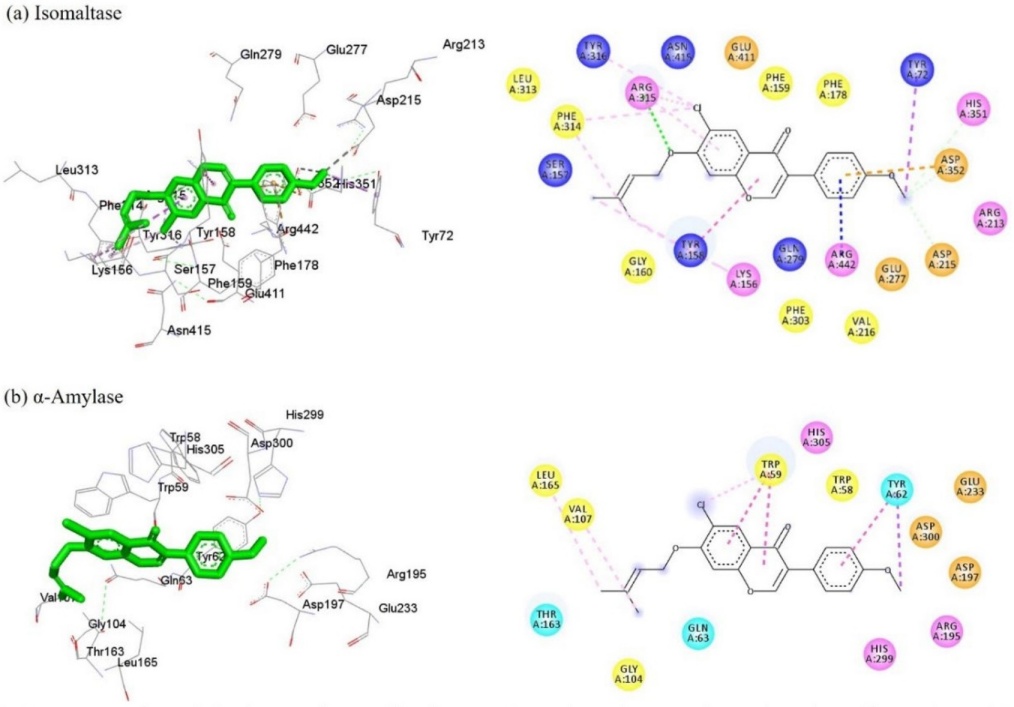
Fig. 3 Representation of the interactions of isoflavone 5c and acarbose at the active site of isomaltase (a) and α-amylase (b). The 3D models illustrate the interactions with the amino acid residues of the catalytic pocket of the enzymes. In the 2D model, conventional hydrogen bond (dark green dotted lines), carbon hydrogen (light green), π-sigma (purple), π-π T-shaped and π-π stacked (fuchsia), π-alkyl and alkyl (pink), π-anion (orange) and π-cation (blue) interactions are shown. The amino acids are depicted with circles of different colors (pink (basic), orange (acid), cyan (polar), and yellow (non-polar)).
For α-amylase, compound 5c exhibiting interactions with some amino acid residues at the site binding pocket of the enzyme, including Trp58, Trp59, Tyr62, Leu165, Asp197, Glu233, and Asp300 [33]. Analysis of docking data showed hydrophobic π-π stacked, π-alkyl, and π-sigma interactions with residues Trp59 and Tyr62. The fragment prenyloxy at C-7 of the isoflavone core is involved a hydrophobic interaction of type alkyl (Leu165 and Val107). This analysis shows that 5c does not interact with the amino acids of the catalytic triad (Asp197, Glu233, and Asp300), suggesting that it is exerting competitive inhibition by interacting at an allosteric site close to the catalytic site.
Lead-likeness, ADME and toxicity prediction
Pharmacokinetics and toxicity predictions of the main α-glucosidase and α-amylase inhibitors 3a, 4a, 5a, and 5c were performed by online software PreADMET and are shown in Table 3 [28]. All title compounds followed Lipinski’s Rule of five and showed moderate permeability to Caco-2 cell. Likewise, these compounds have high human intestinal absorption (HIA). Permeability to blood brain barrier (BBB) and skin for all the title compounds is in the acceptable range. Compound 4a show to be non-mutagenic. Moreover, compounds 4a and 5c have not carcinogenic effect on mouse and rat while compounds 3a and 5a had carcinogenic effect on rat and did not have this on mouse. Furthermore, cardiotoxicity (hERG inhibition) is of medium risk for all the title compounds.
Table 3 Prediction of the lead-likeness and ADMET of compounds 3a, 4a, 5a, 5c, and acarbose.
| Druglikeness/ ADMET a | Compound | ||||
| 3a | 4a | 5a | 5c | Acarbose | |
| Rule of five b | Suitable | Suitable | Suitable | Suitable | Violated |
| Caco2 | 39.0976 | 29.4715 | 41.3766 | 53.5303 | 9.44448 |
| HIA | 97.520337 | 95.737762 | 97.520337 | 97.658677 | 0.000000 |
| BBB | 0.0385324 | 0.342734 | 0.0776034 | 0.17198 | 0.0271005 |
| Skin permeability | -2.5087 | -2.03321 | -2.62531 | -2.6785 | -5.17615 |
| Ames test | Mutagen | Non-mutagen | Mutagen | Mutagen | Non-mutagen |
| Carcino mouse | Negative | Negative | Negative | Negative | Positive |
| Carcino rat | Positive | Negative | Positive | Negative | Negative |
| hERG inhibition | Medium risk | Medium risk | Medium risk | Medium risk | Ambiguous |
a The recommended ranges for Caco2: ˂25 poor, >500 greater, HIA: >80% is high ˂25% is poor, BBB = -3.0 to 1.2, and Skin Permeability = -8.0 to -1.
b MW molecular weight (˂500 g/mol), Log P octanol/water partition coefficient (˂5), Log S aqueous solubility, PSA topological polar surface area, HA hydrogen bond acceptor (˂10), HD hydrogen bond donor (˂5).
Conclusions
Prenylated isoflavones 3a-c and 5a-c were synthesized and their α-glucosidase and α-amylase inhibitory activities were evaluated. Compounds 5a and 5c showed higher α-glucosidase activities and moderate α-amylase activities than standard drug acarbose (6). This suggests that bearing a prenyloxy moiety favors the inhibitory effect against α-glucosidase in comparison to α-amylase. Enzymatic kinetics showed that 5c is a competitive inhibitor for both α-glucosidase and α-amylase enzymes. Docking studies showed that the hydrophobic effect of the prenyloxy moiety at the C-7 position of the isoflavone backbone favors the interaction with both α-glucosidase and α-amylase active sites. Finally, prediction of the lead-likeness and ADMET studies suggest that compounds 5a and 5c are candidate for development of dual inhibitors on carbohydrate-hydrolyzing enzymes.

