











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The Asymmetric synthesis is an important topic in organic chemistry with many applications in the pharmaceutical field. The creation of asymmetric centers by stereoselective synthesis is one of the most important methodologies in asymmetric chemistry. In this context, the asymmetric versions of the aldol addition reaction continue to attract the attention of organic chemists and have been subject of intense synthetic and mechanistic study, because of their importance in the asymmetric construction of carbon-carbon bonds.
It is well known that 1,3-oxazolidin-2-ones 1,[1] 1,3-thiazolidine-2-thiones 2,[2] and 1,3-oxazolidine-2-thiones 3,[3] have been extensively applied as chiral auxiliaries in numerous asymmetric syntheses (Fig. 1).[4]
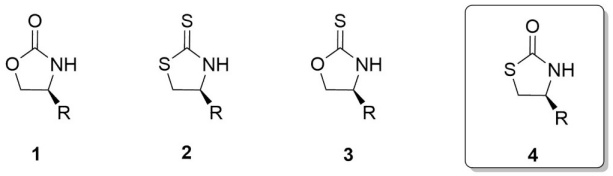
These heterocyclic α-aminoacid-derived chiral auxiliaries have become an integral tool for the preparation of enantiomerically pure intermediates in the synthesis of natural products and specially in the stereoselective preparation of aldol-acetate fragments. Additionally, this class of chiral auxiliaries is popular because of the ease in preparation, their reliable efficiency providing excellent chemical yield and predictable diastereoselectivity.[5]
Evans and co-workers pioneered the use of 1,3-oxazolidinones 1 in asymmetric aldol additions in the early 1980s.[6] These induction-asymmetric reactions resulted in high levels of diastereoselectivity. It was reported that the boron enolates (or their synthetic equivalents) of N-propionyloxazolidinones, undergo aldol addition to aldehydes and ketones in a highly stereo regulated fashion to form the ‘Evans syn’ aldol adduct.[7]
The ‘Evans syn’ aldol adducts, can also be obtained using titanium tetrachloride instead dibutyl boron triflate.[8] In this context, titanium tetrachloride is more economical, and does not require the extra step of oxidation needed to allow hydrolysis of the dibutyl borylaldolates. Titanium enolates, as well as other enolates (e.g. lithium, zinc, or tin derivatives), give rise to ‘non-Evans syn’ aldol adducts when an additional Lewis acid is added to coordinate the aldehyde.[9]
The N-acyl-1,3-oxazolidinethiones 3 were first used as chiral auxiliaries in aldol-type reactions by Nagao and Fujita.[10] More recently, chlorotitanium enolates of N-propionyl 1,3-oxazolidine-2-thiones and N-propionyl-1,3-thiazolidine-2-thione, have been used to access both ‘Evans syn’ and ‘non-Evans syn’ aldol adducts from the same enantiomer of the chiral auxiliary; simply by altering the reaction conditions.[11] Specifically, one equivalent of titanium tetrachloride, (-)-sparteine as the base, and in the presence of N-methyl-2-pyrrolidinone (NMP), gives ‘Evans syn’ aldol adducts with selectivities in ratio of 97:3 to > 99:1. The ‘non-Evans syn’ aldol adducts can also be obtained using 1,3-oxazolidine-2-thiones 3 and 1,3-thiazolidinethiones 2 by altering the stoichiometry of titanium tetrachloride, without NMP present.
On the other hand, the 1,4-addition of nucleophiles to α,β-unsaturated carbonyl systems is one of the most useful methods for asymmetric carbon-carbon or carbon-heteroatom bond formation.[12] The conjugate addition of organometallic reagents to N-enoyloxazolidinone derivatives, has been employed in the synthesis of natural products.[13] Chiral auxiliary-mediated asymmetric Michael addition reactions have been studied extensively and are now an important and general method for asymmetric carbon-carbon bond formation.[14] The addition of organo-copper reagents to chiral alkenoate derivatives (Michael addition) with oxazolidinone, has provided high diastereoselectivity.[15] Alternatively, enoyl-thiazolidinethiones are advantageous as they are more easily cleavable auxiliaries when compared to oxazolidinones 1 or oxazolidinethiones 3.[16] Moreover, there are reports about the transfer of the exocyclic sulfur atom into N-enoylthiazolidinethiones and N-enoyloxazolidinethiones.[17]
Thiazolidin-2-ones 4 are important compounds in both pharmaceutical and synthetic organic chemistry. They are widely encountered as building blocks in natural products with important pharmacological properties,[18] pesticides,[19] and other compounds with anti-HIV3 and anticancer activitiy.[20] Some synthetic methods for thiazolidin-2-one 4 and substituted thiazolidin-2-ones have been developed. Desulfurization (the S/O-exchange) of thiazolidine-2-thiones 2 is an alternative method for synthesizing the corresponding thiazolidin-2-ones 4 via reactions with oxiranes [21] or hydrogen peroxide.[22] Additionally, 4-substituted thiazolidinones also can be generated (45 % - 85 %) yields, from thiazolidinethiones using bromoethanol in etanol with sodium ethoxide as a base.[23]
Experimental
All chemicals were purchased from Sigma-Aldrich and used without any further purification. Thin-layer chromatography (TLC) was visualized using UV light (254 nm). Products were purified by flash chromatography using silica gel (MN Kieselgel 80; Silica gel 60 A; 0,04-0,063 mm / 230 - 400 mesh ASTM). Nuclear Magnetic Resonance (NMR) spectra were measured on Bruker 300 or Varian 500 MHz instruments. 1H NMR chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (δ = 0.0 ppm) with coupling constants (J) reported in hertz (Hz). 13C NMR signals are reported using 77.0 ppm (CDCl3) as the internal reference. Melting points were measured in (Mel-Temp II) instrument without corrections. The optical rotations were measured on a PerkinElmer polarimeter model 343 and the diastereomeric ratios were determined comparing the crude 1H NMR spectra.
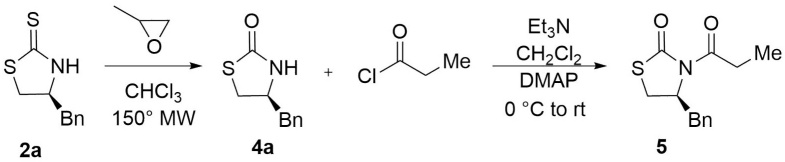
(4S)-Benzyl-1,3-thiazolidin-2-one (4a). Hydrogen peroxide (36 %, 46 µL, 5.47 mmol) was added dropwise to a mixture of (4S)-benzy-1,3-lthiazolidine-2-thione (1.0 g, 4.77 mmol), sodium hydroxide (1.53 g, 38.25 mmol) and water (8.0 mL). The mixture was stirred for 2.0 h at room temperature. Then, aqueous sodium hydrogen sulfite was added, and the desired product was extracted with DCM (3 x 125 mL). The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Purification was accomplished by flash chromatography using silica gel (8:2 hexanes :EtOAc) to afford, after recrystallization from (DCM- hexanes), the (4S)-benzy-1,3-thiazolidine-2-one as a white crystalline solid in 67% yield, mp: 69-71°C, 1H NMR (300 MHz, CDCl3) δ (ppm): 7.39 - 7.20 (m, 3H), 7.23 - 7.13 (m, 2H), 6.07 (s, 1H), 4.07 (p, J = 6.7 Hz, 1H), 3.42 (ddd, J = 11.0, 7.1, 1.7 Hz, 1H), 3.16 (dd, J = 11.0, 6.3 Hz, 1H), 3.01 - 2.83 (m, 2H). 13C NMR (75 MHz, CDCl3) δ (ppm): 174.9 (C=O), 136.5, 129.0, 128.9, 127.2, 56.5 (CH), 40.9 (CH), 34.3 (CH2). EIMS m/z (%): m/z 355 (6, M+), 117 (100). [α]25 D +32.64 (c = 0.01, CHCl3). CCDC 1839074
(4S)-Benzyl-3-propionyl-1,3-thiazolidin-2-one (5). Compound 4a (1.0 g, 5.18 mmol) was dissolved in DCM (10 mL) and cooled to 0 °C. Then Et3N (2.46 g, 24.38 mmol) was added dropwise and stirred for 20 min. Then, propionyl chloride (0.67mL, 0.630 g, 6.85 mmol) was added dropwise and stirred at 0 °C for 1.0 h. The reaction mixture was allowed to reach room temperature and stirred for 2.0 h. The reaction was quenched by the addition of a saturated solution of NH4Cl (15 mL). After further extraction with DCM (3 x 30 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification was accomplished by flash chromatography using silica gel (95:5 hexanes:EtOAc) to afford, after crystallization from (DCM- hexanes), (4S)-benzyl-3-propiony-1,3-thiazolidin-2-one as a white crystalline solid in 61 % yield, mp: 68-70°C, 1H RMN (CDCl3 -300 MHz.) δ (ppm): 7.40 - 7.22 (m, 5H), 4.97 - 4.84 (m, 1H), 3.33 (dd, J = -11.4, 7.4 Hz, 1H), 3.15 (dd, J = -13.2, 3.6 Hz, 1H), 3.08 - 2.74 (m, 4H), 1.17 (td, J = 7.3, 1.8 Hz, 3H). 13C RMN (CDCl3 -300 MHz.) δ (ppm): 173.4 (C=O), 172.3 (C=O), 136.7, 129.4, 128.9, 128.8, 127.1, 77.4, 59.4 (CH), 37.3 (CH2), 30.4 (CH2), 28.6 (CH2), 8.3 (CH3). EIMS m/z 249 (48, M+), 91 (100). [α]25 D +63.7 (c = 0.1, CHCl3).
General aldol condensation procedure
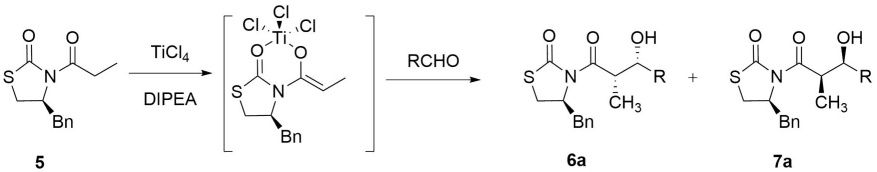
A solution of 5 (100 mg, 0.401 mmol, 1 eq) in DCM (3.0 mL) was cooled at -10 °C, and TiCl4 (60 µL, 1.0 M/ DCM, 1.5 eq) was added dropwise to give an orange solution. After stirring for 20 min at -10 °C, DIPEA (11 µL, 0.805 mmol, 1.5 eq.) was added dropwise. When the reaction turned red stirring continued for 30 min at 10 °C. Then, a solution of the corresponding aldehyde (1.5 eq.) dissolved in DCM (1.0 mmol/mL) was added dropwise. The resulting mixture was stirred for 12 h at -15 °C and then allowed to reach room temperature. The reaction mixture was quenched by the addition of saturated solution of NH4Cl (15 mL) and the desired product was extracted with DCM (3 x 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The diastereoisomeric mixture was purified by silica gel flash chromatography to afford the corresponding aldols 6 and 7 as mixture.
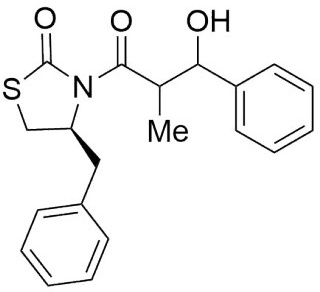
4S)-Benzyl-3-(3-hydroxy-2-methyl-3-phenylpropanoyl) thiazolidin-2-one (6,7a). Following the general procedure, this compound was prepared by reacting 5 with benzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6a and 7a (97:3) as a solid in 86 % yield. After crystallization (DCM-hexanes) mp: 87-89 °C; 1H NMR (300 MHz, CDCl3) δ (ppm): 7.43 (d, J = 7.1 Hz, 2H), 7.39 - 7.22 (m, 8H), 5.16 (s, 1H), 4.96 (ddd, J = 10.6, 7.2, 3.9 Hz, 1H), 4.06 (qd, J = 7.0, 3.4 Hz, 1H), 3.40 - 3.30 (m, 1H), 3.04 (dd, J = -13.2, 4.0 Hz, 1H), 2.94 (s, 1H), 2.93 - 2.83 (m, 2H), 1.19 (d, J = 6.8 Hz, 3H), 1.11 (d, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 175.9 (C=O), 172.4 (C=O), 141.3, 136.4, 130.0, 129.2, 128.9, 128.7, 128.4, 128.3, 128.3, 128.1, 128.0, 127.2, 127.0, 126.1, 126.0, 73.1 (CH), 59.5 (CH), 45.2 (CH), 37.4 (CH2), 28.6 (CH2), 10.5 (CH3). EIMS m/z (%): m/z 355 (6, M+), 117 (100). [α]25 D +32.64 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839075.
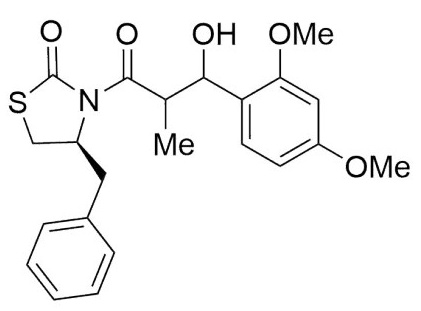
(4S)-Benzyl-3-(3-(2,4-dimethoxyphenyl)-3-hydroxy-2-methyl propanoyl) thiazolidin-2-one (6,7b). Following the general procedure, this compound was prepared by reacting 5 with 2,4-methoxybenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6b and 7b (68:32) as a solid in 86 % yield. After crystallization (DCM- hexanes) mp: 131-133°C; 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38 - 7.30 (m, 4H), 7.30 - 7.21 (m, 6H), 7.07 (d, J = 2.9 Hz, 1H), 6.96 (dd, J = 9.2, 2.7 Hz, 1H), 6.85 - 6.74 (m, 4H), 5.26 (d, J = 4.0 Hz, 1H), 5.07 - 5.01 (m, 1H), 4.97-4.94 (m, 1H), 4.90 (ddd, J = 10.7, 7.4, 3.5 Hz, 1H), 4.45 (p, J = 7.0 1H), 4.25 (qd, J = 7.0, 3.2 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H), 3.74 (s, 3H), 3.28 (dd, J = 11.5, 7.6 Hz, 1H), 3.16 - 3.06 (m, 1H), 3.05 - 2.97 (m, 1H), 2.97 - 2.77 (m, 4H), 1.18 (d, J = 7.3, 3H), 1.06 (d, J = 7.0, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 176.7(C=O), 176.3(C=O), 176.1(C=O), 172.8(C=O), 153.5, 153.4, 153.3, 151.0, 150.4, 150.3, 136.7, 136.5, 136.4, 130.7, 130.1, 129.9, 129.3, 129.3, 129.2, 128.7, 128.7, 127.0, 126.9, 114.5, 114.2, 114.0, 113.9, 113.2, 113.0, 112.9, 112.8, 111.7, 111.3, 111.3, 111.0, (CH), 72.4(CH), 70.4(CH), 59.8(CH), 59.4(CH), 55.7(OMe), 55.6(OMe), 55.6(OMe), 55.6(OMe), 43.8(CH), 43.0(CH), 37.3(CH2), 37.2(CH2), 28.5(CH2), 28.2(CH2), 15.0(Me), 11.7(Me). EIMS m/z (%):415 (74, M+), 166 (100). [α]25 D +38.23 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839076.
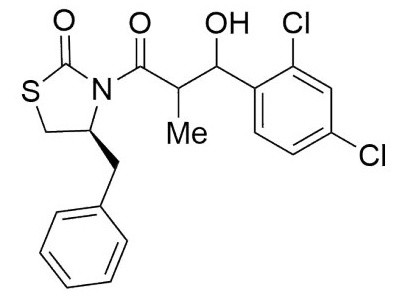
(4S)-Benzyl-3-(3-(2,4-dichlorophenyl)-3-hydroxy-2-methyl propanoyl) thiazolidin-2-one (6,7c). Following the general procedure, this compound was prepared by reacting 5 with 2,4-dichlorobenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6c and 7c (85:15) as a solid in 75 % yield. After crystallization (DCM- hexanes) mp: 151-153°C; 1H NMR (500 MHz, CDCl3) δ (ppm): 7.57 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 7.41 - 7.22 (m, 9H), 5.41 (t, J = 2.2 Hz, 1H), 5.30 (t, J = 7.7 Hz, 1H), 4.94 (ddd, J = 10.6, 7.1, 4.0 Hz, 1H), 4.27 (p, J = 7.0 Hz, 1H), 4.13 (qd, J = 7.2, 2.3 Hz, 1H), 3.84 (d, J = 2.1 Hz, 1H, OH), 3.59 (d, J = 7.6 Hz, 1H, OH), 3.36 (ddd, J = -11.5, 7.3, 1.0 Hz, 1H), 3.18 (dd, J = -13.3, 4.0 Hz, 1H), 3.10 (dd, J = -13.4, 3.5 Hz, 1H), 3.05 - 2.85 (m, 3H), 1.14 (d, J = 7.0 Hz, 3H), 1.06 (d, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): EIMS m/z (%): 428 (1, M + 4), 426(2, M + 2), 424 (3, M+), 117 (100). [α]25 D +11.26 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839077.
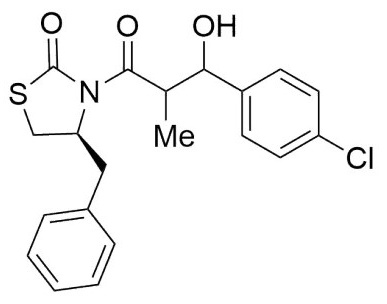
(4S)-Benzyl-3-(3-(2-chlorophenyl)-3-hydroxy-2-methylpropanoyl) thiazolidin-2-one: (6,7d). Following the general procedure, this compound was prepared by reacting 5 with 2-chlorobenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6d and 7d (73:17) as a solid in 77% yield. After crystallization from (DCM- hexanes) it had mp: 176-178°C; 1H NMR (500 MHz, CDCl3) δ (ppm): 7.63 (dd, J = 7.6, 1.9 Hz, 1H), 7.57 (dd, J = 7.6, 1.8 Hz, 1H), 7.40 - 7.17 (m, 12H), 5.48 (t, J = 2.4 Hz, 1H), 5.35 (t, J = 7.6 Hz, 1H), 4.94 (ddd, J = 10.7, 7.2, 3.9 Hz, 2H), 4.33 (p, J = 7.1 Hz, 1H), 4.17 (qd, J = 7.1, 2.4 Hz, 1H), 3.74 (d, J = 2.4 Hz, 1H), 3.51 (d, J = 7.6 Hz, 1H), 3.41 - 3.29 (m, 2H), 3.21 - 3.05 (m, 2H), 3.04 - 2.82 (m, 3H), 1.15 (d, J = 6.9 Hz, 3H), 1.09 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm): 177.3 (C=O), 175.7 (C=O), 173.0 (C=O), 171.9 (C=O), 139.5, 138.2, 136.5, 136.4, 136.3, 133.0, 131.6, 130.8, 129.4, 129.4, 129.4, 129.3, 129.3, 128.9, 128.9, 128.9, 128.8, 128.7, 128.5, 128.5, 128.0, 127.2, 127.2, 127.2, 127.1, 126.6, 126.5, 73.3 (CH), 69.8 (CH), 59.8 (CH), 59.5 (CH), 44.7 (CH), 42.1 (CH), 37.5 (CH2), 37.3 (CH2), 28.5 (CH2), 28.3 (CH2), 14.7 (Me), 10.5 (Me). EIMS m/z (%): 392 (1, M + 2), 390 (2, M+), 57 (100). [α]25 D +78.78 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839078.
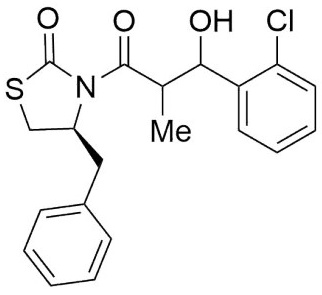
(4S)-Benzyl-3-(3-(3,4-bis (benzyloxy)phenyl)-3-hydroxy-2-methylpropanoyl) thiazolidin-2-one (6,7e). Following the general procedure, this compound was prepared by reacting 5 with piperonal. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (97:3) as eluents, affording a mixture of 6e and 7e (94:6) as an oil in 76 % yield. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.50 - 7.20 (m, 17H), 7.12 - 7.07 (m, 1H), 6.89 (t, J = 1.7 Hz, 2H), 5.20 - 5.14 (m, 2H), 5.11 (d, J = 2.4 Hz, 2H), 5.03 (dd, J = 3.8, 2.2 Hz, 1H), 4.92 (ddd, J = 10.6, 7.3, 3.8 Hz, 1H), 4.05 - 3.95 (m, 1H), 3.38 - 3.23 (m, 1H), 2.98 (dd, J = -13.3, 3.7 Hz, 1H), 2.94 - 2.71 (m, 4H), 1.16 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm): 175.8 (C=O), 172.3 (C=O), 148.7, 148.2, 137.3, 137.2, 136.5, 134.8, 129.4, 128.8, 128.4, 128.4, 127.7, 127.7, 127.5, 127.2, 127.2, 119.2, 114.8, 113.1, 73.0 (CH), 71.4 (OCH2Bn), 71.1 (OCH2Bn), 59.4 (CH), 45.2 (CH), 37.4 (CH2), 28.5 (CH2), 10.7 (Me).. EIMS m/z (%): 549 (1, M+), 43 (100). [α]25 D +10.16 (c = 0.01 g/100 mL, CHCl3).
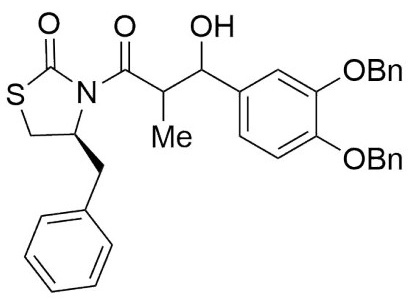
(4S)-Benzyl-3-(3-hydroxy-2-methyl-3-(4-nitrophenyl) propanoyl) thiazolidin-2-one (6,7f). Following the general procedure, this compound was prepared by reacting 5 with 4-nitrobenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (92:8) as eluents, affording a mixture of 6f and 7f (97:3) as a solid in 92 % yield. After crystallization (DCM- hexanes) mp: 150-152°C; 1H NMR (300 MHz, CDCl3) δ (ppm): 8.26 - 8.15 (m, 2H), 7.67 - 7.58 (m, 2H), 7.41 - 7.22 (m, 6H), 5.25 (d, J = 2.7 Hz, 1H), 4.99 (ddd, J = 10.3, 7.1, 4.1 Hz, 1H), 4.02 (qd, J = 7.1, 2.7 Hz, 1H), 3.46 - 3.32 (m, 1H), 3.25 (t, J = 2.7 Hz, 1H), 3.17 (dd, J = -13.2, 4.1 Hz, 1H), 3.06 - 2.92 (m, 2H), 1.13 (d, J = 6.9 Hz, 3H), 1.01 (d, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 175.5 (C=O), 172.8 (C=O), 148.6, 147.2, 136.3, 129.4, 128.9, 127.3, 126.9, 123.4, 72.0 (CH), 59.6 (CH), 44.8 (CH), 37.5 (CH2), 28.9 (CH2), 9.7 (Me). EIMS m/z (%): m/z 401 (3, M+), 102 (100). [α]25 D +14.90 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839079.
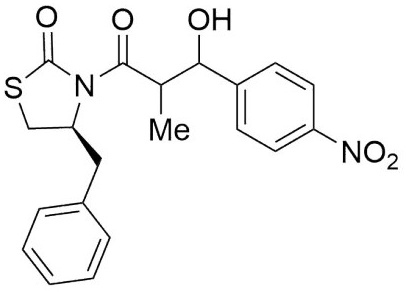
(4S)-Benzyl-3-(3-(4-chlorophenyl)-3-hydroxy-2-methylpropanoyl) thiazolidin-2-one (6,7g). Following the general procedure, this compound was prepared by reacting 5 with 4-chlorobenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6g and 7g (93:7) as a solid in 70 % yield. After crystallization from (DCM- hexanes) it had mp: 124-126°C; 1H NMR (300 MHz, CDCl3) δ (ppm): 7.39 - 7.22 (m, 13H), 5.12 (d, J = 3.1 Hz, 1H), 5.02 (d, J = 3.9 Hz, 1H), 4.95 (ddd, J = 10.7, 7.1, 4.0 Hz, 1H), 4.63 (s, 1H), 3.98 (qd, J = 7.0, 3.1 Hz, 1H), 3.34 (dd, J = -11.5, 7.3 Hz, 1H), 3.08 (dd, J = -13.2, 4.0 Hz, 1H), 2.98 - 2.83 (m, 4H), 1.13 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 175.6 (C=O), 172.6 (C=O), 139.8, 136.3, 133.0, 129.3, 128.8, 128.5, 128.2, 128.2, 127.5, 127.2, 72.3 (CH), 59.5 (CH), 45.0 (CH), 37.4 (CH2), 28.6 (CH2), 10.1 (Me). EIMS m/z (%): 391(10, M + 2), 389 (12, M+), 117 (100). [α]25 D +20.89 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839080.
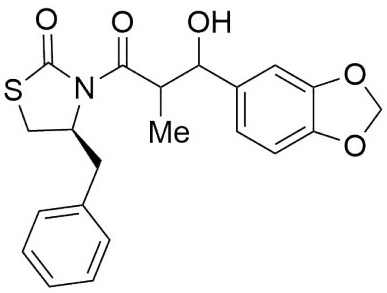
(4S)-Benzyl-3-(3-hydroxy-3-(4-methoxyphenyl)-2-methylpropanoyl) thiazolidin-2-one (6,7h) (Fig. x). Following the general procedure, this compound was prepared by reacting 5 with 4-methoxybenzaldehyde. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (92:8) as eluents, affording a mixture of 6h and 7h (86:14) as an oil in 74 % yield. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.38 - 7.21 (m, 9H), 6.89 - 6.83 (m, 2H), 5.08 (d, J = 3.8 Hz, 1H), 4.99 - 4.88 (m, 1H), 4.72 (d, J = 8.7 Hz, 1H), 4.28 - 4.14 (m, 1H), 4.04 (qd, J = 7.0, 3.8 Hz, 1H), 3.78 (s, 3H), 3.77 (s, 3H), 3.32 (ddd, J = -11.4, 7.3, 1.0 Hz, 1H), 2.99 (dd, J = -13.3, 3.9 Hz, 1H), 2.94 - 2.78 (m, 3H), 1.11 (d, J = 7.0 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm): 175.7 (C=O), 172.4 (C=O), 158.8, 136.5, 136.5, 133.5, 132.2, 129.3, 128.8, 127.9, 127.3, 127.3, 127.1, 113.5, 73.0 (CH), 59.4 (CH), 55.2 (OMe), 45.3 (CH), 37.3 (CH2), 28.4 (CH2), 10.7 (Me). EIMS m/z (%): 385 (M+), 367 (5, M -18) 117 (100). [α]D +24.48 (c = 0.01 g/100 mL, CHCl3). CCDC: 1839081.
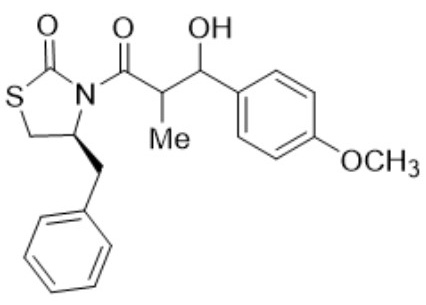
(S)-3-(3-(Benzo[d][1,3]dioxol-5-yl)-3-hydroxy-2-methylpropanoyl)-4-benzylthiazolidin-2-one (6,7i). Following the general procedure, this compound was prepared by reacting 5 with piperonal. The diastereoisomeric mixture was purified by column chromatography using hexanes:EtOAc (95:5) as eluents, affording a mixture of 6i and 7i (97:3) as a solid in 84 % yield. After crystallization from (DCM - hexanes) it had m.p.:103-105°C; 1H NMR (500 MHz, CDCl3) δ (ppm): 7.38 - 7.32 (m, 2H), 7.27 (t, J = 6.3 Hz, 3H), 6.95 (d, J = 1.9 Hz, 1H), 6.89 (dd, J = 8.1, 1.9 Hz, 1H), 6.77 (dd, J = 8.1, 1.9 Hz, 1H), 5.97 - 5.88 (m, 2H), 5.06 (t, J = 2.7 Hz, 1H), 5.01 - 4.90 (m, 1H), 4.03 - 3.95 (m, 1H), 3.39 - 3.28 (m, 1H), 3.13 - 2.96 (m, 1H), 2.97 - 2.76 (m, 4H), 1.19 (d, J = 6.7 Hz, 3H), 1.11 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, 13C NMR (126 MHz, CDCl3) δ (ppm): 175.7 (C=O), 172.4 (C=O), 147.4, 146.6, 136.4, 135.4, 129.3, 128.8, 127.1, 119.3, 107.9, 106.8, 100.8 (CH2), 72.8 (CH), 59.4 (CH), 45.3 (CH), 37.4 (CH2), 28.5 (CH2), 10.5 (Me).EIMS m/z (%): 399 (3, M+), 117 (100). [α]25 D +23.36° (c = 0.01 g/100 mL, CHCl3).
Chiral auxiliary removal
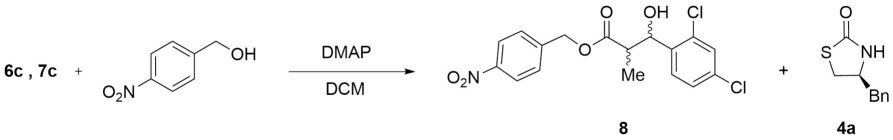
Mixture of compounds 6c and 7c (100 mg, 0.234 mmol) was dissolved in DCM (3.0 mL), p-nitrobenzyl alcohol (40 mg, 0.280 mmol) and 5 mg of DMAP were then added. The reaction mixture was stirred at room temperature for 60 min and quenched by the addition of saturated solution of NH4Cl (15 mL). The crude product was extracted with DCM (3 x 30 mL) and the organic fraction was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification was accomplished by flash chromatography (9:1 hexanes:EtOAc) to give the stereoisomeric mixture 4-nitrobenzyl-3-(2,4-dichlorophenyl)-3-hydroxy-2-methylpropanoate (8) as a yellow oil in a 93 % of yield. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.33 - 8.13 (m, 2H), 7.60 - 7.45 (m, 3H), 7.42 - 7.25 (m, 2H), 5.51 (s, 1H), 5.28 (s, 2H), 3.16 - 2.93 (m, 2H), 1.11 (d, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm): 175.2 (C=O), 147.8, 142.8, 136.9, 133.9, 131.9, 129.3, 129.2, 128.3, 127.1, 123.9, 69.7 (CH), 65.1(CH2), 42.8 (CH), 29.7 (CH), 9.8 (Me).
Results and discussion
In 2012, we reported the synthesis of some derivatives of 1,3-thiazolin-2-ones 4, through desulfurization-oxygenation reaction of 1,3-thiazolidine-2-thiones 2.[24] To our knowledge, no reports dealing with the use of 1,3-thiazolidin-2-ones 4 in asymmetric synthesis have been published so far. In order to explore the synthetic potential and the influence of endocyclic sulfur atom on the chiral 1,3-thiazolidine-2-ones 4, in relation to the application in asymmetric synthesis, we report our preliminary results about the use of these chiral auxiliaries in asymmetric aldol coupling with aldehydes.
Due to the similarity in structure with their congeners 1, 2 and 3, we reasoned that thiazolidine-2-one 4 derivatives, might have applications in the field as well. Therefore, we decided to investigate whether these entities 4 could function as chiral auxiliaries in processes of asymmetric aldol coupling.
We started with the synthesis of (4S)-benzyl-1,3-thiazolidin-2-one 4a[20] from 2a which was prepared according to published procedure from L-phenylalanine.[25] The N-acylation reaction of 4a was achieved using Et3N as base and propionyl chloride as acylating agent, which under these conditions the N-propionyl derivative was obtained in 67 % yield (Scheme 1).
To confirm the absolute structure and absolute configuration of 4a and 5 a single crystal X-ray diffraction was carried out, the compounds 4a and 5 crystalize in the orthorhombic system in the P2(1)2(1)2(1) space group, and the method used for checking the enantiopurity was the Flack parameter,[27] For valid absolute structure assignments, the absolute value of Flack parameter should be close to zero and for enantio-pure compounds s.u. should be less than 0.1. In the case of 4a and 5, the Flack parameters were 0.053(17), 0.02(9) respectively confirming the optical structure of the analyzed samples.
We decided to study the coupling reaction of N-propionyl-(4S)-benzylthiazolidin-2-one 5 with benzaldehyde as a model reaction. Initially, thiazolidin-2-one 5 was treated with LDA (1.0 equiv) in anhydrous THF solution at -78 °C, followed by the addition of benzaldehyde; under these conditions only the cleavage of the chiral auxiliary was observed by TLC. Therefore, we decided try the enolate generation of 5 with TiCl4 (1.0 equiv) in the presence of a base more selective to form aldol adducts as DIPEA (1.0 equiv) at -10 °C (Scheme 2).[26] The analysis by TLC shows a clean reaction with the formation of aldol derivatives 6a:7a in 55 % (chemical yield) as mixture of diastereoisomers with a diastereoselectivity ratio 6a:7a of 82:18. Increasing the amount of Lewis acid, DIPEA and benzaldehyde (1.5 equiv, each/1.0 equiv 5), an improved chemical yield was obtained (86 %), and the diastereoselectivity was observed whit a ratio 6a:7a of 97:3. (Table 1, entry 1). The kinetic diastereomeric ratios were directly determined by comparing the crude 1H NMR spectra.
Table 1 Aldol coupling of thiazolidinone 5 with arylaldehydes.
| Entry a | Aldehyde (RCHO) | 6:7 b | Yield % c |
| 1 | a R = C6H5 | 97:3 | 86 |
| 2 | b R = 2,4-(CH3O)2C6H3 | 97:3 | 84 |
| 3 | c R = R = 2,4-Cl2C6H3 | 85:15 | 75 |
| 4 | d R = 2-ClC6H4 | 73:27 | 77 |
| 5 | e R = 3,4-(BnO)2C6H3 | 94:6 | 76 |
| 6 | f R = 4-NO2C6H4 | 97:3 | 92 |
| 7 | g R = 4-ClC6H4 | 97:3 | 70 |
| 8 | h R = 4-CH3OC6H4 | 86:14 | 74 |
| 9 | i R = 3,4-(OCH2O)C6H3 | 97:3 | 84 |
a For a representative procedure, see Supporting Information.
b Diastereomeric ratios were determined by 1HNMR from mix of diastereomers.
c Yield of the mix of diastereoisomers after purification.
With the optimized reaction conditions in hand, this methodology of asymmetric aldol coupling was extended. We decided probe these reaction conditions with a variety of aldehydes including aliphatic and aromatic derivatives with electron-withdrawing and electron-releasing groups (Table 1).
Important aspects of this asymmetric aldol coupling deserve comment. Firstly, the chemical yield was acceptable in all cases (70 - 92 %), we observed high diastereoselectivity ratio, when a variety of aromatic aldehydes were used independently of the electron nature of the substituent group (electron-releasing or with electron-withdrawing substituent). (See supporting information).
After carrying out the asymmetric aldol condensation to obtain 6 and 7 derivatives, the absolute configuration and the stereochemistry of the new chiral centers created must be verified. This was possible by growing suitable crystals of the major diastereomers (for the compounds 6a, 6c, 6d, 6f, and 6g) and suitable crystals for the diastereoisomeric mixture of 6b and 7b. To these crystals, a single crystal X-ray diffraction study was developed (see support information). The compounds 6a, 6c, and 6d, crystallized in the monoclinic system, in the P2(1) chiral space group. The Flack parameters obtained for the compounds 6a, 6c and 6d were 0.026(9), 0.009(17), 0.039(15) and 0.065(5) respectively. The compounds 6f and 6g crystallized in the orthorhombic system, in the P2(1)2(1)2(1) chiral space group. The Flack parameters obtained for the compound 6g was -0.03(2). The mixture of 6b and 7b crystallized in the monoclinic system, in the P2(1) chiral space group with a Flack parameter of 0.005(4). The configuration was found for the chiral carbons: for the carbon bonded to the benzyl group (C4), for carbon in the alkyl chain bonded to the methyl group (C14), and for carbon in the alkyl chain bonded to the hydroxyl group (C16), in compounds 6a, 6c, 6d, 6f, and 6g, were S, R, R for all cases. In the case of the analyzed crystal of the mixture 6b and 7b, in the asymmetric unit the compounds interact through hydrogen-bonds forming a ring described in graph set terms as
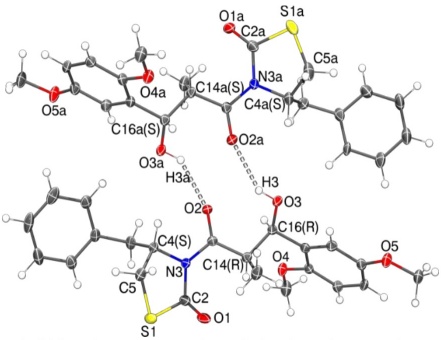
Fig. 2 ORTEP plot at 50 % of probability for asymmetric unit in the mixture of compound 7b at the top and compound 6b at the bottom.
Unfortunately, single crystals for 6e, and 6i suitable for X-ray diffraction analysis could not be obtained and the exact stereochemistry for these compounds was not determined. However, their 1H NMR respective show signals with the same characteristics in chemical shift and coupling constants that the others diastereoisomers 6 and 7. (See supporting information)
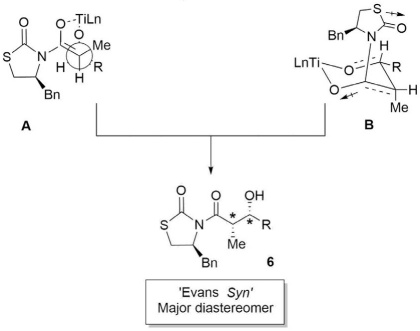
According to the results of the single-crystal X-ray structural analysis, the enolization of N-propanoylthiazolidinone with titanium tetrachloride followed by the coupling reaction with the respective aldehydes at -10 °C, produced the ‘Evans syn’ adduct 6. Probably this can be attributed to a proposed highly non-chelated transition state pre-reacting (TSs) complex A is envisaged first, where the C=O group of the incoming electrophile (benzaldehyde) coordinates with the titanium atom. Coordination from the carbonyl oxygen of the enolate and the electrophile results in a hexacoordinate titanium complex B (Fig. 3). The number of possible conformers for such complexes is therefore restricted. A schematic diagram representing important non-chelated TSs for C-C bond formation between titanium Z-enolate, and benzaldehyde is provided in Fig. 3. The non-chelated TSs, minimizes the dipole interactions between the carbonyl group of the chiral auxiliary and the carbonyl group of the aldehyde along with the developing carbonyl of the aldol group in the thiazolidinone, like oxazolidinones due to presence of the carbonyl group, and dipole minimization considerations could be more important under these reaction conditions.[29] In the non-chelated TS model, attack by the less hindered re face of the enolate on the si face of benzaldehyde is found to be energetically the most favored approach, led to formation of Evans-syn aldol adduct.
Finally, according with the literature this kind of aldol products formed can be converted to a variety of derivatives.[15a] One example was carried out for demonstrate the easy remotion of chiral auxiliary under mild conditions, using the diastereoisomers mixture of 6c:7c, trough the acyl substitution reaction (Scheme 3) with p-nitrobenzyl alcohol in DCM and DMAP 5% mol at room temperature, obtaining the corresponding mixture of stereoisomers 8 in 93 %, yield.
Conclusions
In summary, we have developed a reaction using (4S)-benzyl-1,3-thiazolidin-2-one as a new type of a-aminoacid-derived chiral auxiliary, which provides high levels of diastereoselective in aldol reactions with a variety of aromatic aldehydes in good yields.
A considerable selectivity was obtained for the ‘Evans syn’ aldol product through a non-chelated transition state.
Removal of the chiral auxiliary was carried out under mild reaction conditions to obtain the expected product in a good yield. This kind of derivatives can be easily deprotected due to the presence of the endocyclic sulfur atom in the chiral auxiliary structure.

