











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Peutz-Jeghers (SPJ) o también denominado hipermelanosis mucocutánea es un síndrome genético de herencia autosómica dominante con específicas manifestaciones bucales, cutáneas y gastrointestinales. Dentro de las manifestaciones bucales y dermatológicas se encuentran abundantes máculas melánicas que aparecen en piel perioral, bermellón labial, la totalidad de la mucosa bucal así como en las palmas de las manos y plantas de los pies, mientras que el desarrollo de pólipos hamartomatosos en el tracto digestivo es la principal manifestación gastrointestinal.
Las manifestaciones clínicas de este síndrome fueron descritas por primera vez en 1895 por JRT Conner; sin embargo, no fue hasta 1921 que Peutz vinculó las pigmentaciones mucocutáneas con la poliposis gastrointestinal y más tarde, en 1949 Harold Joseph Jeghers describió de forma conjunta las características clínicas que configuran esta entidad, recibiendo oficialmente el nombre de síndrome de Peutz-Jeghers (SPJ) en 1954.1
El objetivo de este artículo consiste en describir las manifestaciones clínicas bucales y sistémicas del SPJ mediante la exposición de un caso clínico y revisión de la literatura.
Revisión de la literatura
Manifestaciones gastrointestinales
La presencia de pólipos gastrointestinales es la manifestación clínica más frecuente en el SPJ. Se caracterizan por lesiones hamartomatosas producidas a expensas de músculo liso, lo cual le da a los pólipos una apariencia circunvolucionada. De hecho, la característica distintiva de los pólipos encontrados en el SPJ es que son pedunculados y presentan un patrón de arborización de fibras de músculo liso proveniente de la muscularis mucosae que se extiende al interior del pólipo.1,2
Histopatológicamente los pólipos se consideran hamartomas por tratarse de tumores de naturaleza benigna cuyas células tienen apariencia normal, pero se encuentran desorganizadas con respecto a la arquitectura tisular del sitio donde aparecen. Los pólipos presentan epitelio columnar absortivo y células de Paneth, estas últimas permiten diferenciar un pólipo hamartomatoso propio del SPJ de un pólipo adenomatoso intestinal. La mayoría de estos pólipos se caracterizan también por tener una abundante actividad mitótica epitelial, pero sin actividad displásica.1-3
A pesar de que los pólipos pueden aparecer en cualquier parte del sistema digestivo, desde la unión gastroesofágica hasta el canal anal, se ha observado que el yeyuno (65%) y el íleon (55%) son los sitios más frecuentes de aparición;4 sin embargo, también pueden presentarse en recto (36%), estómago (23%) y duodeno (15%).5
Asimismo, los pólipos se vinculan fuertemente con intususcepción y obstrucción intestinal,3-5 condición que puede llevar a la muerte si no es tratada quirúrgicamente de forma oportuna.
Si bien la mayoría de los pólipos del SPJ se consideran lesiones hamartomatosas benignas, otros tienden a malignizar, sobre todo aquéllos que se desarrollan a nivel del duodeno, estómago y colon,6 y por tal motivo, su extirpación mediante técnicas endoscópicas y quirúrgicas para su análisis histopatológico siempre es obligatorio para esclarecer esta distinción.
Manifestaciones mucocutáneas
Las manifestaciones mucocutáneas propias del SPJ, conocidas como melanoplaquias, son un conjunto de máculas hipermelánicas con un diámetro menor de 5 mm cuya coloración puede ser azulada o negruzca, pueden ser únicas o múltiples que confluyen entre sí. Topográficamente aparecen en la piel adyacente a orificios anatómicos como piel peribucal y mucosa bucal, piel periorbitaria, perianal, perigenital y piel de las palmas de las manos y plantas de los pies.4-7
La zona más frecuente de aparición son los labios y la mucosa de revestimiento (mucosa labial y yugal), pudiendo encontrarse también a lo largo de la mucosa masticatoria (paladar duro y encías). La lengua y la mucosa del piso de boca raramente se ven afectadas.
La melanoplaquia no es un hallazgo congénito, pues no está presente al nacimiento, y suele aparecer en los primeros dos años de vida. Son signos de suma importancia para el diagnóstico del SPJ, ya que la aparición de estas pigmentaciones melánicas en piel y mucosa bucal puede ser el motivo de consulta en pacientes pediátricos no diagnosticados.
Se ha descrito en la literatura que la melanoplaquia tiende a despigmentar la piel con el paso del tiempo durante la adolescencia o la adultez; no obstante, las hiperpigmentaciones en mucosa oral son permanentes.6,8,9
Histológicamente, las máculas bucales presentan acantosis y melanina en la capa basal con melanocitos con procesos dendríticos elongados.7
Diagnóstico diferencial
Si bien las manifestaciones propias del SPJ son claras y el diagnóstico de esta condición parece no ser complejo, no debe dejarse de lado que existen otras enfermedades y síndromes donde la poliposis gastrointestinal y la hipermelanosis mucocutánea son frecuentes y por tanto, deben tomarse en cuenta toda vez que se sospeche que un paciente padece SPJ.
Otras enfermedades que pueden producir pigmentaciones mucocutáneas son:
Enfermedad de Addison caracterizada por insuficiencia corticosuprarrenal primaria, dentro de sus manifestaciones mucocutáneas se encuentran la hiperpigmentación de piel y mucosas debida al déficit de glucocorticoides y a la hormona estimulante de melanocitos.10
Síndrome de Laugier Hunziker que presenta máculas hiperpigmentadas en piel y mucosa oral, frecuentemente vinculado con hiperpigmentación ungueal.11
Síndrome de Carney es una enfermedad autosómica dominante caracterizada por mixomas, manchas mucocutáneas e hiperactividad endócrina.12
Síndrome de McCune Albright que presenta manchas cutáneas color café con leche, displasias fibrosas de hueso y pubertad precoz.13
Resultados:Síndrome de Cowden es una enfermedad genética cutánea caracterizada por manifestaciones en piel y mucosas y una fuerte predisposición a desarrollar hamartomas que pueden malignizar.14
Asimismo, existen otros síndromes genéticos que si bien no presentan pigmentaciones mucocutáneas, muestran una alta predisposición a desarrollar pólipos gastrointestinales; entre los más comunes destacan el síndrome de Bannayan-Riley-Ruvalcaba, el síndrome de Gardner y la poliposis adenomatosa familiar.15
Caso clínico
Se presenta el caso de un paciente de 11 años con diagnóstico de síndrome de Peutz-Jeghers diagnosticado a los tres años de edad tras la aparición de múltiples máculas encontradas de forma generalizada en piel y mucosa oral. Al ingreso del paciente, se realiza el llenado de la documentación médica de rutina que incluye historial médico, exploración clínica de cabeza y cuello así como consentimiento informado y aviso de privacidad donde se detallan los alcances de la información personal y elementos fotográficos recabados.
A referir de la madre, el médico pediatra alertó del SPJ por las manifestaciones mucocutáneas que presentaba el paciente, se interconsultó con médico genetista quien confirmó el diagnóstico a través de las manifestaciones clínicas apoyándose en una prueba de cariotipo. Se realizó además una exhaustiva exploración a los padres del menor sin encontrar antecedentes del SPJ en la familia, motivo por el cual se determinó que se trataba de una mutación de novo.
Dentro de los antecedentes personales patológicos del paciente, se encuentra antecedente de dos pólipos intestinales en intestino delgado diagnosticados a los ocho años de edad mediante estudios de radiografía con medio de contraste y tratados mediante resección quirúrgica de los mismos.
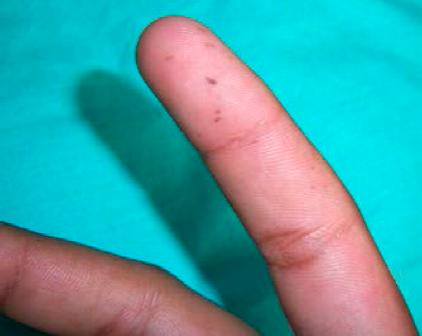
A la exploración clínica se observan múltiples máculas que confluyen entre sí en el borde bermellón del labio superior e inferior, siendo más abundantes en el labio inferior (Figura 1). Presenta también máculas generalizadas en mucosa labial y mucosa yugal (Figura 2), y sólo unas cuantas a nivel de la papila incisiva y paladar duro (Figura 3), respetando la mucosa lingual y piso de boca. El paciente presentó una mancha melánica difusa en la falange distal del dedo índice de la mano derecha (Figura 4).
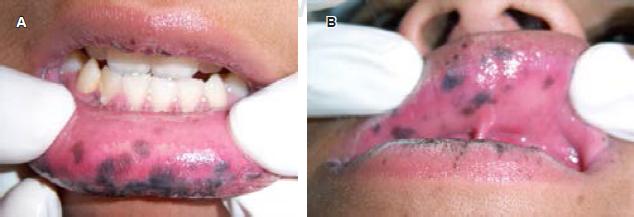
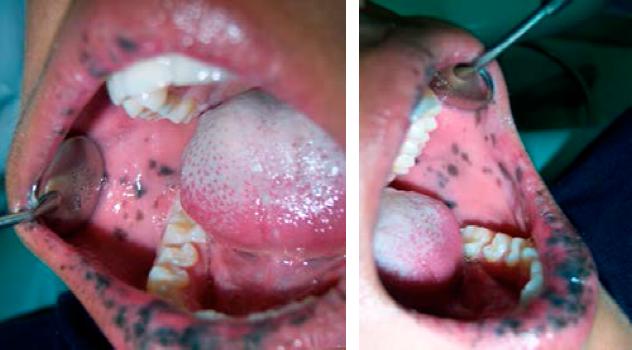
Figura 2 Hipermelanosis en la mucosa yugal y comisuras de forma bilateral, en estas imágenes se puede observar que se encuentran intactas la mucosa lingual y el piso de boca.
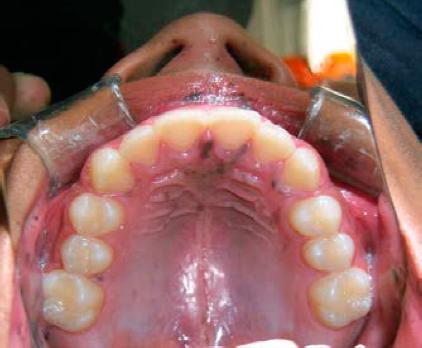
Figura 3 Máculas dizfusas en paladar duro predominando a nivel de la papila incisiva. Nótese que también presenta manchas melánicas adyacente a las fosas nasales.
Discusión
El SPJ es una condición que se transmite vía autosómica dominante por una alteración en el brazo corto del cromosoma 19, y puede ser transmitida a 50% de la descendencia en ambos sexos, es decir, que cuando uno de los padres es el afectado, corre el riesgo de 50% en cada embarazo de procrear un hijo(a) afectado y 50% de que nazca sano.
Sin embargo, en el presente caso se trata de una mutación de novo, lo que significa que la mutación en el gen afectado apareció por primera vez en la familia por no existir antecedentes familiares del síndrome.
Si bien el SPJ se asocia, como ya se mencionó, con hipermelanosis mucocutánea y poliposis gastrointestinal, también se han reportado pólipos nasales, quistes ováricos, tumores de Brenner, disgerminomas, tumores de células de Sertoli, cáncer de mama y adenocarcinomas pancreáticos. En 1895 la Sociedad Científica de Londres presentó el caso de dos hermanas gemelas con SPJ, reportando el fallecimiento de ambas a causa de obstrucción intestinal y cáncer de mama respectivamente,4 lo cual evidencia que la interrelación del SPJ con otros subtipos de padecimientos es un hecho conocido desde hace más de un siglo. En 1921 Peutz estudió de cerca una familia holandesa que presentaba pigmentaciones y pólipos gastrointestinales en varios miembros de la familia, el pedigree de la familia mostró que más de la mitad de sus miembros desarrollaron cáncer intestinal.16
Un dato importante que mencionar es que los pólipos presentados durante la infancia son totalmente benignos, y su carácter maligno se desarrolla hasta la adultez.8
Como ya se mencionó con anterioridad, las máculas bucales pueden no estar presentes al nacimiento y aparecer con el paso del tiempo durante la infancia. En algunos pacientes ancianos se ha observado que las máculas cutáneas tienden a despigmentarse con el paso de los años, pero no así las máculas bucales. No obstante, pese a la despigmentación de las máculas cutáneas no se disminuye el riesgo de desarrollar pólipos gastrointestinales.5
No existe evidencia de que las pigmentaciones hipermelánicas malignicen como los pólipos gastrointestinales.8
Conclusión
El SPJ es un trastorno genético autosómico dominante que afecta a hombres y mujeres por igual, y cuyas manifestaciones clínicas visibles se encuentran en su mayoría confinadas a la mucosa bucal y piel peribucal. Resulta imposible separar la genética de la odontología, dada la alta correlación que existe entre los síndromes genéticos y las manifestaciones craneomaxilofaciales que éstos presentan, y por ello el odontólogo debe desarrollar la destreza para detectar condiciones que se apartan de la normalidad y que a su vez puedan ser sugestivas de alguna condición genética que no haya sido diagnosticada. El diagnóstico clínico y genético oportuno del SPJ permite al profesional de la salud alertar al paciente sobre las posibles comorbilidades que pudieran acontecer a lo largo de su vida como el riesgo de sufrir intususcepción, oclusión o cáncer gastrointestinal.

