










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista odontológica mexicana
versión impresa ISSN 1870-199X
Rev. Odont. Mex vol.15 no.3 Ciudad de México jul./sep. 2011
Case report
Adams-Oliver syndrome. Case report
Mariana Godínez Barragán,* Gustavo Parés Vidrio,§ Alejandro Hinojosa Aguirre,II Adolfo Yamamoto Nagano¶
* Pedodontics student at DePel, School of Dentistry, National University of Mexico.
§ Tutor. Pedodontics Specialty, School of Dentistry, National University of Mexico.
II A Consultant. Pedodontics Professor at the School of Dentistry, National University of Mexico.
¶ Consultant, Pedodontics Specialty Coordinator DePel, School of Dentistry, National University of Mexico.
Received: 31 July 2009.
Accepted: 2 July 2010.
ABSTRACT
Adams Oliver syndrome (AOS) is a rare disease characterized by congenital scalp defects, terminal transverse limb malformations, and cutis marmorata telangiectatica, whose treatment requires a multidisciplinary focus to improve the patient s quality of life. We report the case of a child afflicted with varied manifestations of AOS including craniofacial alterations in the form of facial fissure, bilateral cleft lip and grade III cleft palate. Therapeutic focus of this case was approached in the dental area, taking into account the patient s systemic conditions. The process was divided into five phases taking into consideration the patient s current age, as well as intraoral and craniofacial circumstances.
Key words: Adams Oliver syndrome, aplasia cutis congenita terminal transverse limb defects.
INTRODUCTION
Aplasia cutis congenita (ACC) is an heterogeneous group of alterations characterized by congenital absence of epidermis, dermis and occasionally hypodermis. Its most frequent location is the scalp.1,2 Although Cordon1-4 described the first ACC case in 1767, it is Campbell' report in 1826 which allowed to differentiate its clinical aspects.
According to Frieden1-5 lesions were classified into nine groups according to location, inherency mode and associated anomalies. Within these groups there have been reports of patients with alterations manifested as distal reduction of the limbs, this embodies a clinical combination which represents an ACC group II with dominant autosomal inherency and variability in the clinical expression, called Adams Oliver Syndrome (AOS).6,7 The first report of AOS was published in 1945 by Forrest H Adams and CP liver.8-11 AOS is a rare congenital alteration showing an incidence of 200 world cases, according to the last record published in 2004. This, according to Martinez Frias's12 Estudio Colaboración Español de Malformaciones Congénitas (Collaboratiave Spanish Study of Congenital Malformations) represents a frequency of 0.44 for every 100,000 live births. Male-female gender distribution is similar according to research conducted by Whitley13 in 1991 on patients located in one geographic area.
In Mexico, the first AOS case was presented by Barcelà14 at the 2004 Congreso Nacional de Genética Humana (National Congress of Human Genetics). Nevertheless, the first report in medical literature pertains to Lujan8 a physician of the Juarez Hospital in Mexico City. AOS represents a varied phenotype whose main components include congenital absence of skin in the form of aplasia cutis congenita, defects in transverse terminal portions of limbs, and hereditary cutis marmorata telangiectasica in 25% of cases. ACC is found in the posterior zone of the parietal or vertex region. It manifests itself as lesions which vary between 0.5 and 10 cm with dilated veins and defects in the parietal bones.11 Defects in the transverse terminal portions of limbs are malformations in the shape of distal phalanx truncations which propitiate their absence. There has been report of cases with syndactyly (bone/skin) between the second and third fingers, proximal and medial phalanx reduction, polydactyly, ectrodactyly, metatarsus hypoplasia, nail hypoplasia, club foot, absence of the distal section of the member, micromyelia and brachipodia.
Other rare anomalies include optical nerve hypoplasia, microftalmia,16 eye alterations,17 vitreum and retina malformations18 in the shape of sickle shaped folds, congenital bilateral cataracts,20 defects in the ear pavillions skin appendages in the toes, ACC of the knee, focal hyperpigmentations, hemangiomas, artery-vein malformations of the scalp, woolly hair, cryptorchidism, polythelia, growth stunt,20,21 mental retardation, short physical size, liver sclerosis,22 intrahepatic anomalies22 portal fibrosis22 dolichocephaly and cleft palate.8,23,24 According to Roca's report, the AOS cases with clefts represent a rate of 0.3%.25
AOS syndrome has been associated to neurological anomalies of the central nervous system like meningitis secondary to infections caused by skin defects, encephalitis,26 porencephalic cysts, asymmetric cerebral hypoplasia of the callous body18,19,28 intracranial calcifications of the TORCH2 type, cerebral cortex dysplasia29 hydrocephaly28 and psychomotor retardation.12
In AOS patients, a 13.4% incidence of cardiac malformations are reported. Hereditary (congenital) cardiopathies are mentioned like aortic coarctaction and ventricular and atrial septum defects30 subaortic and aortic stenosis aortic coarctation, pulmonary vein stenosis, Fallot31 tetralogy, atresia, uncommonly shaped mitral valve, bicuspid aortic valve and pulmonary atresia.
There is one case associated with chronic myeloid leukemia and chylothorax.32
AOS cases present an enormous scope of clinical diversity, ranging from major malformations through to minimal alterations, therefore showing a varied scope of expressions.9
AOS is reported most frequently as dominant autosomal inherence with pronounced variability. These data are supported by authors such as Bonafede and Beighton6 in 1979, Sybert6 in 1989 and Verdych33 in 2006. Some publications have suggested recessive hereditary traits, like Kahn and Olmedo in 1950, Koiffmann in 1988, Klinger and Merlob in 1998, Tekin in 1999 or Temtamy17 in 2007. Nevertheless, sporadic cases have been described due to novo mutations8,9,15 incomplete penetration in the transmission mode9 or spontaneous genetic changes such as those described by Narang.34 For Becker,35 family consanguinity, incomplete chromosome penetrability or mosaicism can determine the type of heritage. Based on the aforementioned, researchers like Verdyck33,36 have tried to decipher genes implicated in AOS development, like ALX4,36 MSX2,36 MSX1,33 CART1,33 RUNX2,33 HODXD1333 and P63. These attempts have not yet met with success, for this reason it has been linked to pathological and physiological processes. Toriello9,23,37 suggests the presence of vascular disruptive mechanisms in early embryonic stages, the same as Whitley and Gorlin13 who suggested a vascular compromise in watershed areas during development periods. Jaeggi,38 Keymolen39 and Fryns40 inform that this process is the one which best describes AOS. Swartz41 suggests that vascular anomalies are developed in very early stages of embryonic development.
Der Kaloustian42 deducted that AOS and Poland s disease resulted from subclavian artery interrupted blood flow, and therefore the same chromosome alteration. This possibility has not been verified.
Other hypotheses include rupture of amniotic bands39 teratogenic factors, intrauterine infections43 like chickenpox, zoster or simple herpes, fetal exposure to cocaine, heroin, alcohol or antithyroid drugs43 oligohydramniosis and external compressions9 but nothing has been substantiated to that effect.
The great clinical heterogeneity of AOS affected patients has hindered genetic counsel to anomaly carriers. It represents a medical challenge which requires a multidisciplinary approach to attain appropriate valuation, prevention, management and control.
This study has the objective of sharing a new case in the Mexican Literature with a multidisciplinary approach, to relate it with different medical areas involved in its treatment and to draw attention on the importance of pedodontics in such a process.
CLINICAL CASE
The patient was a male infant, 1 year 9 months of age, taken to the Pedodontics Clinic of the Graduate and Research Division of the School of Dentistry, National University of Mexico, to improve his dental and oral condition.
Medical history
Medical history revealed that the patient was the product of a first pregnancy of a 20 year old mother and 22 year old father. Upon interrogation, parents denied any prior family hereditary problems. The mother experienced a pregnancy of normal evolution. Upon prenatal regular controls, at 28 weeks of intrauterine life, an increase of the fetus encephalic contour and anomalies in development of upper and lower limbs were detected.
The child was born after 40 weeks of pregnancy, through a Caesarian Section and was diagnosed with hydrocephaly. At birth the child weighed 3,700 g, had encephalic contour of 42 cm and a ¾ degree of Apgar depression. Physical examination showed craniofacial deformities such as bilateral cleft lip, cleft palate, facial cleft of left palpebral corner to the left wing of the nose, eye anomalies, and enlargement of cranial veins. The patient also suffered of malformations of terminal transverse segments of the limbs manifested as right and left club feet, finger malformations of the left hand with syndactyly between second and third phalanx, nail hypoplasia, reduction of proximal and medial phalanges, and syndactyly of the right foot. In accordance with clinical characteristics present at birth, the patient was diagnosed with AOS.
The patient remained in hospital for the first days of his life, where he was operated to receive a unidirectional (ventricle-peritoneal) valve to control hydrocephaly. Cleft lip and facial cleft were corrected. The right clubfoot was also corrected and the joined fingers of the left hand were separated. There were no complications. Additionally the patient was evaluated in order to determine psychomotor development. To obtain this evaluation the Denver test was used, which, when the patient failed over two tests, determined psychomotor deficiency as consequence of AOS upon exploring fine and broad motor skills , personal social area and language. From that point onwards, and up to the present, the patient underwent stimulation programs to enhance the following fields; visual, propioceptive auditory, bodily image and stabilization of the four limbs. A series of exercises were set up to increase encephalic control and muscular tone. The patient experienced evident improvement of development.
When the patient reached 1 year and 18 days of age, he was again operated to correct the left clubfoot. The operation was totally uneventful.
The patient was the object of frequent clinical evaluations and controls until the age of 1 year and 4 months (Figure 1).
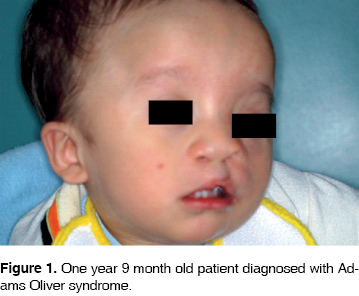
Physical exploration (examination)
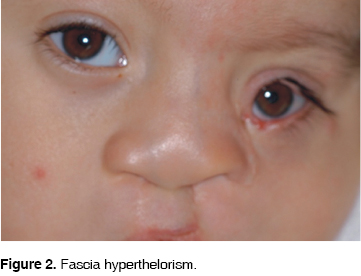
Physical exams were carried out at the Pedodontics Clinic of the DePeI (Graduate Studies and Research Division) Faculty of Dentistry, National University of Mexico. A diagnosis of dolichocephaly was emitted with superciliary planes, both papillary subnasal and corner planes anti parallel, a vertical to horizontal nasal relation of 1 to 1 with asymmetric and elongated orifices, as consequence of the presence of the scar of the bilateral cleft lip operation and the surgery of the facial oblique cleft which projects downwards the left side of the face. The upper lip is thin with pronounced asymmetry up to the vermillion border which is directed to the left side by the scar of the bilateral cleft lip operation. The fascia is hypertheloric (Figure 2) with low implantation of the external ear.
Terminal transverse defects were also observed in the left arm in the form of syndactyly in the second and third phalanx where a scar can be observed. Phalanges of the right leg are hypoplastic and absence of nail bed is observed. Both legs show scars from club foot corrective surgery (Figure 3).
Oral and dental diagnosis
The diagnosis of the mouth was divided into three sections:
1. Intraoral examination. Lips presented dehydrated mucosa, there was a poor match between upper and lower lip due to the scars of the cleft surgery; soft and hard palate were cleft up to the incisive orifice according to Veau (Figure 4), tonsils were swollen and the irregular alveolar process in the shape of right unilateral cleft in the pre- maxilla area was accompanied by a collapse in the zone. There was also circumscribed gingival swelling due to the presence of bacterial plaque.
2. Occlusion analysis: Occlusion analysis was not undertaken in view of the dental age of the patient (14 to 18 months).
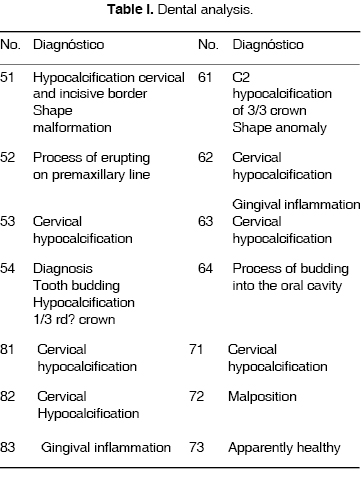
3. Dental analysis (Table I).
Treatment plan
The patient presented several alterations associated with AOS. A multidisciplinary approach taking into account medical, psychological, social and pedagogical aspects was deemed basic to improve the patient s quality of life. Specialists who should take part in this comprehensive treatment are pediatricians, geneticists, otolaryngologists plastic surgeons, anaesthetists neurologists, orthopedists maxillofacial surgeons, pedodontists, orthodontists audiologists and speech therapists, family psychologists, physiotherapists, nutritionists and social workers. Oral and dental treatment was divided into different phases according to the current age of the patient. These included:
Determination of behavior management technique: The age of the patient precluded communication. There were also AOS related anomalies like eye alterations, psychomotor deficiencies, delay in growth and alterations of the NCS. Therefore, physical approach techniques were used, like the use of mouth openers and passive immobilization technique, to protect physical integrity in the patient and reduce parental anxiety. Nevertheless in major procedures, the use of anaesthesia must also be considered in view of the general and neurological conditions of the patient.
Prevention phase
In this phase instructions were given on hygiene of the palatine cleft, alveolar process and the tongue. Use of a gauze or clean cloth soaked in boiled water or diluted peroxide was recommended. The importance of this procedure, especially after the last meal of the day, was emphasized.
Parents were instructed in brushing techniques, with a soft brush made of silicon or latex, without toothpaste, to be implemented after meals. Any doubts on hygiene were cleared at this point. Parents were made to understand the importance of improving the mouth to counteract hypocalcified zones present in most teeth of the patient, since deformities in the distal portions of the patients limbs, can in the future interfere in his performing proper hygiene practices by himself.
A low carbohydrate diet as well as dispensing with nocturnal feeding was recommended.
At a later appointment a professional dental prophylaxis was performed, having previously observed an antibiotic course. This was so decided since the patient presented high risk of developing meningitis, septicemia or endocarditis since he was afflicted with a one directional ventricle-peritoneum valve, therefore in concordance with the American Heart Association44 Amoxicillin, 50 mg/kg was administered 60 minutes before initiating the procedure. After prophylaxis with toothpaste and rubber cup, fluoride varnish was applied.
Restorative phase
To restore caries in tooth 61, a traumatic restorative technique was employed, eliminating softened tissue with a spoon and low speed handpiece. Glass ionomer Type II was used as restorative material.
Interceptive phase
Since the patient presented grade III cleft palate, there was the possibility to trigger a series of functional and structural problems which had to be timely intercepted. It was therefore recommended to contemplate maxillofacial surgery to jointly design a presurgical orthopedic device to decrease segments and facilitate fissure closure, normalize lingual pressure during the process of deglutition and suction, and form an artificial palate to avoid the lodging of the tongue in the fissure and allow the normal translation of both segments.
The patient has to be evaluated for this palate surgery during this interceptive stage.
Once the closure of the clef is attained, it is necessary to reorient and promote maxilla and mandibular growth to avoid secondary effects to the scar flanges. The use of skeletal expansion, disjunction and traction with customized orthopaedic devices in order to obtain appropriate transverse, vertical and anteroposterior relations can be considered.
During this postsurgical stage the intervention of audiologists and speech therapists will be of utmost importance to avoid speech and hearing alterations.
Maintenance phase
This phase is very important to ensure orthodontic and dental treatment success. In this phase, evaluation is made of advances sustained in oral hygiene, diet changes, occlusal development, craniofacial growth and proper relation between maxilla and mandible.
DISCUSSION
AOS represents an extremely rare congenital alteration of ACC. Its clinical manifestations and mode of inheritance are so varied that each report in scientific literature shows the diversity of cases the clinician might have to face.
Medical advances must focus on finding the gene responsible for its development, to then be able to understand pathological and physiological developments which in turn cause vascular anomalies, and the interruption of blood flow. This in turn triggers defects in the distal sections of limbs of all patients reported as afflicted with SAO. If it is possible to solve this enigma, early diagnosis will be of crucial value. Becker35 mentions this when he stresses the importance of constant evaluation during the first three months of pregnancy, using fetoscopy35 to discard this or any other syndrome which affects fetal development. In our days, it is not recommended to use prenatal procedures like amniocentesis5,23 or the study of chorionic villi5,23 to diagnose AOS, since results would show a normal karyotype5 and an alteration free chromosome analysis. The clinical operator must therefore base his diagnosis on clinical findings and family history, and so be able to discard syndromes which come accompanied of ACC of the vertex like syndrome of skin defects of the vertex and postaxial polydactyly, syndrome of vertex skin defects with hands as clamped hands , bullosa epidermolysis, ectodermal dysplasia, Johanssen Brizzard syndrome, embryofetopathy caused by congenital chickenpox, 13 trisomy, focal dermal hypoplasia, syndrome of congenital ring finger constriction, and intrauterine amputations due to rupture of amniotic bands.2
AOS management is complex, since there is no specific treatment for the disease. We can only improve the circumstances of the patient, and this multidisciplinary approach is of utmost importance. This approach will change AOS patients circumstances from birth, allowing for a better quality of life.
It is important to point out that mortality in AOS patients is low, it represents 20%8,15 of documented patients. The main cause for this mortality were haemorrhages in the aplasia zone, secondary infections and meningitis.8,15,45,46
Dentistry is one of the main sciences involved in the multidisciplinary approach for AOS. According to the patients age and the dental and mouth circumstances, the dentist might devise a program to counteract or prevent alterations. To be able to achieve this, it is important to take into account the great variability of clinical manifestations of AOS. AOS has been linked to hepatic22 anomalies, neurological, vascular and cardiac anomalies in 13% of documented cases,29 therefore, instauration of dental treatments must be devised after proper medical evaluations. Then in AOS patients also afflicted with congenital cardiopathies, or patients with ACC with bone defects which expose portions of the brain, preventive measures like the use of antibiotic prophylaxis are extremely important to avoid endocarditis.
This case is a new finding for Mexican scientific literature. Clinical manifestations of this case, as has happened before, are diverse, but they concur in the presence of distended veins, limb deformities in the form of syndactyly (webbed fingers and toes) proximal and medial phalanx reduction, nail hypoplasia, club foot, eye anomalies, growth hindrance, hydrocephaly, psychomotor delay, facial cleft, bilateral cleft lip and grade III cleft palate, the latter being a rare finding in AOS patients, representing 0.3% of cases.25
The therapeutic access for this case was approached through dental and oral perspectives. At this stage, objectives were to improve oral hygiene circumstances, and they favored the appropriate control of food intake with high carbohydrate content to counteract hypocalcified zones present in teeth. With this we achieved a more favorable oral medium, and established then a lower number of risk factors for the development of carious lesions. With the presence of a cleft palate, a timely surgical intervention will avoid a greater maxillary collapse due to the lack of closure of palatine processes, anomalous position of the tongue during deglutition and suction processes, as well as improved craniofacial relationships.
CONCLUSIONS
• AOS represents a rare congenital alteration, insufficiently documented in scientific literature. This shows the need to document news cases.
• AOS requires of a multidisciplinary approach to achieve better management and control.
• Pedodontics is part of the work team since it offers an improvement in the dental and oral circumstances, it controls risks factors to avoid caries development, as well as deleterious habits or malocclusion.
• Oral and Dental treatment of AOS requires previous multidisciplinary valuations which take into account preventive measures.
• Comprehensive treatment of AOS patients is continuous, from birth until the achievement of the best medical, social and psychological circumstances.
REFERENCES
 Mailing Address:
Mailing Address:
Dr. Gustavo Parés Vidrio
E-mail: drgpares@yahoo.com.mx
Note
This article can be read in its full version in the following page: http://www.medigraphic.com/facultadodontologiaunam
1. Hona J, Frieden MD. Clinical review. Aplasia cutis congenital: A clinical review and proposal for classification. J Am Acad Dermatol 1987; 4 (4): 646-60. [ Links ]
2. Calduch LJR. Aplasia cutis congenital de cuero cabelludo. Estudio clínico de 35 pacientes y su relación con los defectos de la línea media. (Tesis doctoral). Universitat de Valencia. Facultad de Medicina 2005. [ Links ]
3. Cordon M. Extrait d' une lettre au sujet de trois enfants de la meme mere nés avee partie des extremites denuee de peau. J Med Chir Pharmacie 1767; 26: 556-557. [ Links ]
4. Pérez LC, Urbina FG, Roa JA. Aplasia cutis congénita: a propósito de cuatro casos. Rev Chil Pediatr 2001: 72 (4). [ Links ]
5. Jaeggi E, Kind C, Morger R. Congenital scalp and skull defects with terminal transverse limb anomalies (Adams-Oliver syndrome): report of tree additional cases. Eur J Pediatr 1990; 149 (8): 565-6. [ Links ]
6. National Center for Biotechnology Information. Adams-Oliver syndrome. http://www.ncbi.nlm.nig.gov
7. Online Mendel Ian Inheritance in Man. Adams-Oliver Síndrome; AOS. http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=100300
8. Luján JI, Ibarra AG, Méndez FV. Síndrome de Adams-Oliver. Reporte de un caso. Rev Hosp Jua Mex 2004; 71 (3): 124-7. [ Links ]
9. Mempel M, Abeck D, Lange I, Strom K. The wide spectrum of clinical expression in Adams-Oliver syndrome: a report of two cases. British Journal of Dermatology 1999; 140: 1157-1160. [ Links ]
10. Adams FH, Oliver CP. Hereditary deformities in man due to arrested development. J Hered 1945; 36: 3-7. [ Links ]
11. Scrubabu NMD. The syndrome of aplasia cutis congenital with terminal, transverse defect of limbs. Brief clinical and laboratory observations. The journal of Pediatrics 1975; 87 (1): 79-82. [ Links ]
12. Martínez-Frías ML, Arroyo-Carrera I, Muñoz-Delgado NJ. The Adams-Oliver syndrome in Spain: the epidemiological aspects. An Esp Pediatr 1996; 45 (1): 57-61. [ Links ]
13. Whitley CB, Gorlin RJ. Adams-Oliver syndrome revisited. Am J Med Genet 1991; 40 (3): 319-26. [ Links ]
14. Barcela SL. Síndrome de Adams-Oliver. Caso Clínico. Revista Salud Pública y Nutrición. Edición Especial. XXIX Congreso Nacional de Genética Humana 2004. URL: http://www.respyn.uanl.mx/especiales/2005/ee-05-2005/carteles/genetica_clinic_a1.htm [ Links ]
15. Bilginer B, Onal MB, Bahadir S, Akalan N. Aplasia cutis congenital of the scalp, skull and dura associated with Adams-Oliver syndrome. Turkish Neurosurgery 2008; 18 (2): 191-193. [ Links ]
16. Salvador J, Casas J. Catálogo de síndromes polimalformativos congénitos con anomalías oculares. Instituto Municipal de Salud Pública. Ayuntamiento de Barcelona. http://www.aspb.es/quefem/docs/Catal_sind_polimalformativos
17. Temtamy SA, Aglan MS, Ashour AM, Zaki MS. Adams-Oliver syndrome: further evidence of an autosomal recessive variant. Clin Dysmorphol 2007; 16 (3): 141-9. [ Links ]
18. Ortavik KH, Strömme P, Spetalen S, Flage T, Westvik J, Vesterhus P, Skjeldad O. Aplasia cutis congenita associated with limb, eye, and brain anomalies in sibs: a variant of the Adams-Oliver syndrome? Am J Med Genet 1995; 59 (1): 92-5. [ Links ]
19. Prothero J, Nicholl R, Wilson J, Wakeling EL. Aplasia cutis congenital, terminal limb defects and falciform retinal folds: confirmation of a distinct syndrome of vascular disruption. Clin Dysmorphol 2007; 16 (1): 39-41. [ Links ]
20. Fayol L, Garcia P, Denis D, Philip N, Simeoni U. Adams-Oliver syndrome associated with cutis marmorata telangiectatica congenital and congenital cataract: a case report. Am J Perinatol 2006; 23 (39): 197-200. [ Links ]
21. McGoey RR, Lacassie Y. Adams-Oliver syndrome in siblings with central nervous system fingings, epilepsy, and developmental delay: refining the features of a severe autosomal recessive variant. Am J Med Genet A 2008; 146 (4): 488-91. [ Links ]
22. Girard M, Amiel J, Fabre M, Pariente D, Lyonnet S, Jacquemin E. Adams-Oliver syndrome and hepatoportal sclerosis: occasional association or common mechanism? Am J Med Genet A 2005; 135 (2): 186-9. [ Links ]
23. Sun-Young Jun, Shin Kwang Khang. An autopsy case of Adams-Oliver syndrome. J Korean Med Set 2000; 15: 482-4. [ Links ]
24. Rajabian MH, Aghaei SMD. Adams-Oliver syndrome and isolated aplasia cutis congenital in two sibs. Dermatology Online Journal 2001: 12 (6). [ Links ]
25. Roca OJL, Cendán MI, Alonso LF, Ferrero OME, Lantigua CA. Caracterización clínica del labio hendido con fisura palatina o sin ésta en Cuba. Rev Cubana Pediatr 1998: 70 (1). [ Links ]
26. Gomes LB, Castro J, Matos M, Numes A, Furtado J, Barrueco MC. Lesion of the central nervous system in Adams-Oliver's syndrome. Acta Med Port 2001; 14 (1): 89-94. Acta Med Port 2001; 14 (4): 455-6. [ Links ]
27. Bamforth JS, Kaurah P, Byrne J, Ferreir P. Adams Oliver syndrome: a family with extreme variability in clinical expression. Am J Med Genet 1994; 49 (4): 393-6. [ Links ]
28. Piazza AJ, Blackston D, Sola A. A case of Adams-Oliver syndrome with associated and pulmonary involvement: further evidence of vascular pathology? Am J Med Genet A 2004; 130A (2): 172-5. [ Links ]
29. Zapata HH, Sletten LJ, Pierpont ME. Congenital cardiac malformations in Adams-Oliver syndrome. Clin Genet 1995; 47 (2): 80-4. [ Links ]
30. Papadopoulou E, Sifakis S, Raissaki M, Germanakis I, Kalmanti M. Antenatal and postnatal evidence of periventricular leukomalacia as a further indication of vascular disruption in Adams-Oliver syndrome. Am J Med Genet A 2008; 146A (19): 2545-2550. [ Links ]
31. Lin AE, Westgate MN, van der Velde ME, Lacro RV, Holmes LB. Adams-Oliver syndrome associated with cardiovascular malformations. Clin Dysmorphol 1998; 7 (4): 235-41. [ Links ]
32. Farrell SA, Warda LJ, LaFlair P, Szymonowicz W. Adams-Oliver syndrome: a case with juvenile chronic myelogenous leukemia and chylothorax. Am J Med Genet 1993; 47 (8): 1175-9. [ Links ]
33. Verdyck P, Blaumeiser B, Holder-Espinasse M. Adams-Oliver syndrome: clinical description of a four-generation family and exclusion of five candidate genes. Clin Genet 2006; 69: 86-92. [ Links ]
34. Narang T, Kanwr AJ, Dogra S. Adams-Oliver syndrome: a sporadic occurrence with minimal disease expression. Pediatr Dermatol 2008; 25 (1): 115-6. [ Links ]
35. Becker R, Kunze J, Horn D, Gasiorek-Wiens. Autosomal recessive type of Adams-Oliver syndrome: prenatal diagnosis. Ultrasound Obstet Gynecol 2002; 20: 506-510. [ Links ]
36. Verdyck P, Holder-Espinasse M, Van Hull W, Wuyts W. Clinical and molecular analysis of nine families with Adams-Oliver syndrome. European Journal of Human Genetics 2003; 11: 457-463. [ Links ]
37. Toriello HV, Graff RG, Florentine MF. Scalp and limb defects with cutis marmorata telangiectatica congenital: Adams-Oliver syndrome? Am J Med Genet 1988; 29: 269-76. [ Links ]
38. Jaeggi E, Kind C, Morger R. Congenital scalp and skull with terminal transverse limb anomalies (Adams-Oliver syndrome): report of tree additional cases. Eur J Pediatr 1990; 149 (8): 565-6. [ Links ]
39. Keymolen K, De Smet L, Bracke P, Fryns JP. The concurrence of ring constrictions in Adams-Oliver syndrome: additional evidence for vascular disruption as common pathogenetic mechanism. Genet Couns 1999; 10 (3): 295-300. [ Links ]
40. Fryns JP, Legius E, Demaerel P, Van den Berghe H. Congenital scalp defect, distal limb reduction anomalies, right spastic hemiplegia and hipoplasia of the left arteria cerebro media. Further evidence that interruption of early embryonic blood supply may result in Adams-Oliver (plus) syndrome. Clin Genet 1996; 50 (6): 505-9. [ Links ]
41. Swartz EN, Sanatani S, Sandor GG, Schreiber RA. Vascular abnormalities in Adams-Oliver syndrome: a cause or effect? Am J Med Genet 1999; 82 (1): 49-52. [ Links ]
42. Der Kaloustian VM, Hoyme HE, Hogg H, Entin MA, Guttmacher AE. Possible common pathogenetic mechanisms for Poland sequence and Adams-Oliver syndrome. Am J Med Genet 1991; 38 (1): 69-73. [ Links ]
43. Suárez O, López-Gutiérrez JC, Andrés A, Barrera S, Encinas JL, Luis A, Soto-Bauregard C, Días M, Ros Z. Aplasia cutis congenital: surgical treatment and results in 36 cases. Cir Pediatr 2007; 20 (3): 151-5. [ Links ]
44. Rick A, Nishimura, Blase A, Carabello, David P, Faxon, Michael D, Freed, Bruce W Lytle, Patrick T. O'Gara, Robert A, O'Rourke. Guideline update on valvular heart disease: Focused Update on Infective Endocarditis. JACC 2008; 52 (8): 676-85. [ Links ]
45. Savarirayan R, Thompson EM, Abbott KJ, Moore MH. Cerebral cortical dysplasia and digital constriction rings in Adams-Oliver syndrome. Am J Med Genet 1999; 86 (1): 15-9. [ Links ]
46. Davis PM, Buss PW, Simpson BA, Sykes. Near fatal hemorrhage from the superior sagital sinus in Adams-Oliver syndrome. Archives of Disease in Childhood 1993; 68: 433. [ Links ]

