











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Alzheimer's disease (AD) is defined as a type of dementia characterized by gradual and progressive neurodegeneration secondary to neuronal death1. Among the etiologies of dementia, approximately 80% are attributed to AD, making it the most prevalent type worldwide2,3. Trends in AD show a 147.95% increase in its incidence from 1990 to 2019, with 7.29 million cases worldwide in 20194. This makes it one of the most significant chronic pathologies in terms of mortality, quality of life, and public health over the past century5.
AD is a multifactorial condition, with age being the most significant risk factor for its development. The prevalence of AD tends to double every 5 years after the age of 656. According to the Global Burden of Diseases, patients aged 70-74 show the highest incidence of AD. The aging population and increased life expectancy are the main contributors to the rise in the incidence of this pathology in recent years. It is estimated that by 2050, 152 million people will be living with AD worldwide4. The pathophysiology of AD involves environmental, genetic, and lifestyle factors2. While age is the most significant risk factor for AD, other identified risk factors include hypertension, obesity, diabetes mellitus (DM), physical inactivity, hearing loss, smoking, low educational attainment, social isolation, high alcohol consumption, traumatic brain injury, and environmental pollution7.
Despite currently available treatments, AD remains a progressive condition for which there is no prevention or cure. So far, no treatment has advanced from being symptomatic to having an effect on the prognosis of the disease3. This work aims to provide a comprehensive review of the present knowledge on AD, diagnostic methods, treatments, and potential tools under study that show promising results for patients with this disease.
Generalities of AD
Pathophysiology
AD is a progressive neurodegenerative condition mainly affecting older adults, characterized by a decline in cognitive and functional abilities, along with behavioral changes. The disease progresses over 15-25 years, during which patients transition from normal cognitive function to dementia. During the early stages, patients may remain asymptomatic or experience mild to moderate neurocognitive impairment5.
The pathophysiology of AD involves multiple biological processes, with the amyloid-ß (Aß) cascade hypothesis being widely accepted8. Abnormal processing of amyloid precursor protein (APP) leads to the formation of Aß plaques, which accumulate in the brain, causing synaptic dysfunction and neurodegeneration9. In addition, tau protein abnormalities and neurofibrillary tangles are also key pathological features that disrupt neuronal function and further exacerbate disease progression2,8.
GENETIC FACTORS
Genetic factors significantly influence both early and late-onset AD10. Mutations in the APP, presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes contribute to abnormal Aß plaque production, leading to inflammation, neuronal toxicity and death3,5,9,10. These mutations, following an autosomal dominant pattern, are responsible for familial AD, a rare form affecting 0.5% of cases, with symptoms typically appearing between ages 30 and 50. However, not all variants of these gene mutations result in familial AD, some may act as disease modifiers5,9. Among the genes associated with this disease, PSEN1 mutations on chromosome 14 are the most prevalent (71.24%), followed by APP (46.97%), and PSEN2 (20.63%)11.
Another mutation associated with the development of AD is that of apolipoprotein E (ApoE), located on chromosome 19, with three isoforms identified: ε2, ε3, and ε412. The mutation of the ε4 allele of the ApoE gene has been identified as the most significant genetic risk factor for both early and late-onset AD, occurring in approximately 65% of cases1,12. Carriers of one or two copies of the ε4 allele have a higher risk of developing the disease1,10.
INFLAMMATORY PROCESSES IN AD
Inflammatory processes are key in AD development, involving astrocytes, microglial cells, cytokines, and chemokines that trigger neuroinflammation13. This inflammatory response damages the neuronal environment, contributing to oxidative stress and cellular apoptosis, leading to the onset of characteristic AD symptoms14. Neuroinflammation is a specific and enhanced immunological, biochemical, and hematological response that can be triggered locally or systemically. This process is a reaction to tissue or organ damage, designed to neutralize harmful factors and stimulate repair processes15.
The most important substances exerting a pro-inflammatory effect in the brain and the body include cytokines (Interleukin-1 [IL-1ß], IL-6, IL-18, Tumor necrosis factor-α, Interferon-γ), chemokines (CCL2, CCL3, CXCL8), complement components (C1q, C5), transcription factors (NF-κB), peptides (bradykinin), enzymes (COX-2, iNOS, LOX), and coagulation factors platelet activating factor. Conversely, anti-inflammatory effects are exerted by lipoxins (LXA4, RvE1) and some cytokines (IL-10, IL-37, transforming growth factor-ß)12. In classical inflammation, the process is characterized by an increased secretion of pro-inflammatory mediators and a reduction of anti-inflammatory substances. To resolve the inflammatory response, the opposite occurs, pro-inflammatory substances decrease, and anti-inflammatory substances increase. Neuroinflammation in AD differs from peripheral inflammation. In the brain, neuroinflammatory factors, such as cytokines and chemokines can persist longer and display different dynamics compared to those in systemic inflammation16.
Diagnostic methods
The diagnosis of AD is a complex process, requiring the evaluation of the patient's medical history, physical and neurological examinations, neuropsychological tests, and imaging studies. There is no single test that can definitively diagnose AD, therefore, the diagnosis is based on a combination of these different approaches16. Regarding medical history, the physician will review the patient's medical background, including any memory, behavior, or language problems, as well as family history. Neuropsychological tests are used to assess the patient's cognitive abilities, such as memory, attention, language, and problem-solving. These tests can help identify specific areas of cognitive impairment that may be associated with AD17. Imaging tests, such as positron emission tomography and magnetic resonance imaging, can help visualize the brain and detect changes that may be associated with the disease. These tests can help rule out other causes of dementia and determine the severity of the disease18.
Conventional treatment
The treatment approach for AD is symptomatic, as there are currently no curative or prognosis modifier treatments3. Conventional pharmacological treatment focuses on cognitive improvement and the regulation of neuropsychiatric symptoms5. At present, there are two groups of drugs approved for use in AD: Acetylcholinesterase inhibitors and N-methyl-D-aspartate receptor antagonists9.
Relationship between insulin resistance and AD
The global increase in DM and AD has posed a major challenge for people, healthcare systems, and governments worldwide. Conventionally, studied separately due to the peripheral implications of DM and the central implications of AD, the growing prevalence of these conditions has led to a deeper exploration of their interconnected pathophysiology, particularly their shared pro-inflammatory underpinnings19. DM is a systemic, chronic-degenerative disease that is closely related to lifestyle and profoundly affects the quality of life of millions of people worldwide. Expected to reach a prevalence of 10.9% by 2045, DM manifests itself in three main types: Type 1, type 2, and type 3. Type 1 DM (DM1) results from the destruction of pancreatic β-cells, while type 2 DM (DM2) is due to impaired insulin resistance and secretion20. Type 3 DM (DM3) is a relatively new concept characterized by brain insulin resistance caused by the presence of metabolic syndrome and microglial inflammation induced by reactive oxygen species and neurotoxins, leading to neuronal stress and neurodegeneration21.
There are two mechanisms by which DM2 increases the risk of damage to neurons, neuroglia, and the neurovascular unit of the brain: (1) neuronal senescence (complications in patients older than 65 years) and (2) metabolism (hyperglycemia and hyperinsulinemia, increased Aß toxicity, Tau hyperphosphorylation, oxidative stress, and neuroinflammation19,21. This metabolic state may lead to impaired neuronal function and cognitive impairment. This metabolic dysregulation further contributes to the formation of neuritic plaques, hippocampal atrophy, and lower cerebrocortical glucose metabolism, which is closely related to memory decline20,19. Kapogiannis et al. have demonstrated that at elevated levels of p-Ser312IRS1 (a kinase involved in autophagy) in patients with prodromal AD, suggesting that insulin resistance may develop years before clinical symptoms appear22.
In addition, neuronal-derived exosomes have shown promise as potential biomarkers for early diagnosis of AD23. Defective insulin signaling in the brain in AD and DM2 may result from several factors, including insulin resistance, negative regulation of insulin receptors (IR), reduced binding affinity, or faulty activation of the downstream signaling cascade24. Ultimately, this leads to a decrease in neuronal glucose uptake, manifesting as a decline in neuroplasticity, neurotransmitter deficiencies, bioenergetic collapse, and the onset of a detrimental inflammatory cascade. The disruption of insulin signaling alters brain metabolism, potentially paving the way for neurodegeneration and providing a compelling explanation for the link between DM, obesity, and AD23,24.
Insulin and insulin-like growth factors (IGF-1) exert their effects by binding to specific tyrosine (Tyr) kinase receptors, such as the IR and the IGF-1 receptor. While the highest expression of IRs is observed in the olfactory bulb, cerebral cortex, and hippocampus, they are also present in the endothelial cells of the blood-brain barrier (BBB). This specific localization of IRs plays a crucial role in mediating the transport of insulin and IGF-1 across the BBB, thereby influencing the function of the central nervous system (CNS)25.
Pancreatic β cells within the islets of Langerhans produce insulin, its effect by binding to transmembrane glycoprotein receptors composed of α and β subunits. These bindings are facilitated by disulfide bonds within the insulin molecule26. The initial step in insulin signaling involves the binding of the ligand and insulin to the extracellular α subunits of the IR, a transmembrane glycoprotein receptor complex composed of two α subunits and two β subunits. This binding event triggers conformational changes within the receptor, leading to the activation and autophosphorylation of specific Tyr residues located in the cytosolic region of the β subunit27. These phosphorylated residues then serve as binding sites for the substrates of the IR, particularly Insulin receptor substrate 1 (IRS-1) and IRS-2, which act as crucial intermediaries in the propagation of the insulin signal28. The assembly of these IRS-containing complexes orchestrates the initiation of multiple intracellular signaling cascades25. The hyperinsulinemia pathway (a state of excessive circulating insulin) is linked to AD due to the saturation of insulin-degrading enzymes, which hampers their ability to eliminate both insulin and Aß peptides, the latter being a hallmark of AD pathology. The higher incidence of AD in individuals with type DM2 and obesity further strengthens the notion of shared mechanisms29. Notably, although neuronal glucose uptake may not be entirely reliant on insulin, the notion of "brain insulin resistance" underscores the disruption of insulin signaling pathways. This disruption is increasingly recognized as a key factor in the pathogenic pathway between AD and DM223.
New treatments
Antidiabetic pharmacological agents for Alzheimer
Oral hypoglycemic agents that target insulin resistance in the brain by promoting proper incretin and insulin signaling are currently a promising option for the treatment of AD and other neurodegenerative diseases. Glucagon-like peptide-1 (GLP-1) receptor agonists have demonstrated neuroprotective and anti-inflammatory properties, with expression observed in neurons and glial cells30. GLP-1 receptors activate a series of anti-inflammatory pathways by stimulating adenylyl cyclase, which increases cAMP concentrations. This activates other cofactors that have an anti-apoptotic effect. In addition, various genes, such as Bcl-2, are activated, which are neuroprotective and inhibit the activation of pro-apoptotic proteins, such as caspases and p5330,31.
Insulin, a hormone that regulates peripheral glucose homeostasis, also has significant implications for the nervous system. It swiftly crosses the BBB and can be synthesized by glial cells32. Although cerebral glucose metabolism is not dependent on insulin, this hormone can influence glucose concentrations through neuronal glucose transporter 4 in cognitive circuits and enhance glycogen absorption in astrocytes. In addition, insulin modulates neurotransmitter levels, such as dopamine, provides protection against the synaptotoxic effects of Aß, and plays a crucial role in vascular function due to its involvement in lipid metabolism and inflammation32,33. These attributes suggest that insulin may offer a therapeutic alternative for diabetic patients with cognitive impairment or AD.
HYPOGLYCEMIC AGENTS IN THE TREATMENT OF AD
The research on specific drugs for AD treatment remains in its early stages, offering promising potential for reducing the degenerative damage caused by AD. Among the hypoglycemic agents studied are Metformin, the most commonly prescribed antidiabetic drug, and liraglutide33. Zheng et al. carried out a Mendelian randomization study on 527,139 Europeans from the general population, of whom 71,880 were either diagnosed with or at risk of developing AD. The study identified 22 metformin-related genes across 5 targets associated with the glycemic marker hemoglobin A1C, with brain connections confirmed through 6,601 brain donors, showing that 20 of the 22 genes were linked to the cerebral cortex and cognitive function. Results indicated that metformin use reduces the risk of AD by 15% in diabetic individuals and by 4% in healthy individuals34. In a mouse model, metformin was found to promote chaperone-mediated autophagy, a process related to the pathophysiology of neurodegenerative diseases. AD is marked by progressive neuronal loss and Aß peptide oligomerization, resulting in amyloid plaque formation. Metformin has been shown to reduce Aß levels, leading to decreased cognitive impairment, delayed aging, and alleviation of age-related diseases34,35. Another antidiabetic drug that has shown evidence in reducing neurodegeneration is liraglutide, a GLP-1 inhibitor34. This medication may promote the development of stem cell stimulants, potentially leading to the proliferation of new neurons. In mouse models, it has been associated with calorie restriction, which decreases the amount of Aß plaques and neurofibrillary tangles. The use of GLP-1 reduces food intake and hunger. A mouse model study used liraglutide treatment for 2 months and found improvements in spatial memory and a 30%-50% reduction in inflammation36. Peripheral intravenous liraglutide demonstrated stimulation of the insulin Akt signaling pathway, preventing tau phosphorylation and reducing GSK3ß activity in mice37 (Table 1).
Table 1 Mechanisms of action of antidiabetic agents with potential neuroprotective effects in Alzheimers disease
| Drug | Mechanism of action | References |
|---|---|---|
| Metformin | Reduces Aß levels and amyloid plaque formation, decreasing neuronal degeneration and cognitive decline | 35 |
| Liraglutide | Promotes the activity of the insulin signaling pathway Akt, preventing tau phosphorylation. GLP-1 receptors activate a series of anti-inflammatory pathways with anti-apoptotic effects. Various genes, such as Bcl-2, which are neuroprotective, are activated, and pro-apoptotic proteins are inhibited | 30,31,37 |
| Insulin | Modulates neurotransmitter levels such as dopamine, providing protection against the synaptotoxic effects of amyloid-beta protein | 32,33 |
GLP-1: glucagon-like peptide-1; Aß: amyloid-ß. Metformin, liraglutide, and insulin have demonstrated promising effects in preclinical models of Alzheimers disease. These include reduction of amyloid-beta accumulation, anti-inflammatory activity, and modulation of neurotransmitter systems relevant to cognitive function.
Stem cell therapy in AD
The pharmacological treatments currently used for AD have certain limitations in managing the disease, as they show poor response in altering its progression. This has led to the ongoing search for new therapies to improve the quality of life for patients. Since there is currently no cure for AD, only disease-modifying treatments, the use of stem cells has recently been promoted as a therapeutic approach. Specifically, neural stem cells (NSCs), induced pluripotent stem cells (iPSC), and mesenchymal stromal cells (MSC) have shown promise in this area38,39.
NSCs
NSCs are a type of multipotent stem cell, considered primitive cells with the ability to differentiate into various specialized cell types, give rise to other cells, and self-renew. These cells can generate different cell types in the CNS such as glia, neurons, and oligodendrocytes38,40. Due to these properties, NSCs have been investigated for their potential benefits in AD. NSCs are essential for CNS recovery in neurodegenerative diseases associated with oxidative damage, as seen in AD. The mechanism of action of these cells involves identifying the injury site and differentiating into astrocytes, neurons, and oligodendrocytes to adapt and promote cellular plasticity. In addition, they can reduce inflammation41. Endogenous activation or exogenous infusion of NSCs restores neural networks in the brains of AD patients, regenerates damaged areas, and plays a crucial role in cognition and memory. The transplantation of NSCs aims to enhance neural connections, improve the inflammatory environment of the brain secondary to AD, and regenerate the neural network that undergoes degeneration through the secretion of chemokines41. The use of NSCs in vitro has been shown to generate cholinergic neurons that improve memory performance, enhance cognitive function, reduce neuroinflammation, and increase neurogenesis in rodent models of AD39. In addition, the administration of these cells improves the migration, survival, and differentiation of neuronal cells, thereby enhancing memory40. Further clinical studies are needed to assess their efficacy in AD patients.
iPSC
iPSC have been significant since their first mention in 200742, particularly in the analysis of the pathogenesis of diseases influenced by genetic and environmental predispositions, such as age. Their application in neurodegenerative diseases is primarily focused on pathological research and the development of new drugs43. Their continuous differentiation potential into the three germ layers makes iPSCs a valuable source for managing various diseases44. Derived from the somatic cells of patients or healthy individuals, iPSCs have been demonstrated to be reprogrammed and differentiated into brain-specific cells, such as neurons, astrocytes, microglia, oligodendrocytes, pericytes, and vascular endothelial cells45. This capability facilitates the replacement of lost cells, the release of trophic factors and extracellular matrix, and the improvement of the aging environment through neuroprotection and inflammation suppression, ultimately leading to enhancements in neurodegenerative diseases43. This new technology continues to advance not only in the field of AD but also in Parkinson's disease, Amyotrophic Lateral Sclerosis, and Fibrodysplasia Ossificans Progressiva43. Although iPSC models have not yet reached the gold standard, they offer numerous advantages over other models in understanding the pathological mechanisms of AD45. In the case of these diseases, conducting precise analyses in animal models is challenging due to the complexity of recreating pathological phenotypes that accurately mimic neurodegenerative diseases43.
MSC
MSC exhibits typical stem cell characteristics, such as the ability to differentiate into various cell lineages46. They have played a significant role in correcting neurodegenerative disorders due to their active migration to damaged sites (homing), immunomodulatory and neuroprotective effects, as well as their roles in cellular repair and angiogenesis46,47. The ability of MSC to promote tissue repair through paracrine factors such as growth factors and cytokines allows modulation of the pro-inflammatory response, creating a conducive environment for neurogenesis and improving neurological deficits. Therefore, they are recognized as an innovative therapeutic option for inflammatory and chronic degenerative diseases46. MSC are attracted to sites of inflammation and can be obtained from various tissues such as bone marrow, adipose tissue, umbilical cord, amniotic fluid, placenta, and peripheral blood. When administered systemically, they cross the BBB, facilitating the elimination of Aß plaques, promoting neurogenesis, and reducing apoptosis. They also have the capacity to provide a healthy supply of mitochondria to the CNS, thereby mitigating the effects of aging and mitochondrial dysfunction related to AD. These actions can improve neuronal morphology and enhance spatial behavioral memory48,49. Studies in animal models of AD have confirmed the therapeutic potential of MSC. When administered to the brains of mice with AD, MSC secrete cytotropic factors-proteins capable of influencing cell growth and survival-rather than differentiating into neurons and glial cells, which can address multiple pathogenic mechanisms of AD50. In a study by Cone et al. MSC treatment significantly improved mouse memory and decreased the number of Aß plaques in the hippocampus51. In clinical studies, the primary focus is on evaluating the safety and toxicity of treatment doses. While some studies have observed elevated serum levels of biomarkers in control groups, such as vascular endothelial cells, IL-12, IL-10, IL-4, IL-6, and IL-2, which collectively promote an anti-inflammatory environment, transient fever has also been reported after each administration52 (Fig. 1).
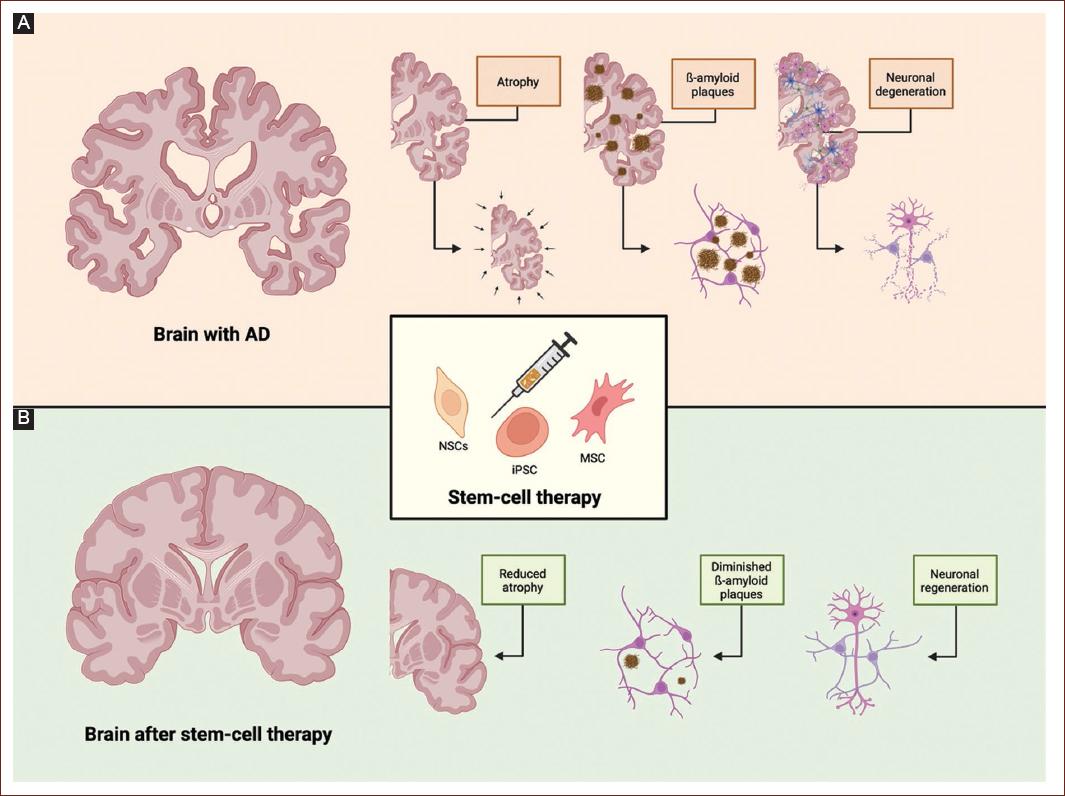
Figure 1 Pathological changes in the brain affected by Alzheimer's disease and the potential reparative effects of stem cell therapy. Panel A illustrates the hallmark neurodegenerative features of Alzheimers disease, including progressive cerebral atrophy, the accumulation of ß-amyloid plaques, and widespread neuronal degeneration, all of which contribute to cognitive decline. These pathological changes are closely linked to elevated levels of neuroinflammatory cytokines and synaptic dysfunction. Panel B shows the potential therapeutic effects of stem cell-based interventions, including the administration of mesenchymal stromal cells (MSCs), induced pluripotent stem cells (iPSCs), and neural stem cells (NSCs). Following treatment, the AD brain demonstrates significant improvements: reduced cortical atrophy, decreased ß-amyloid plaque load, and signs of neuronal regeneration. The stem cell therapy may mediate their neuroprotective effects through several mechanisms: (i) differentiation into neural cell types, (ii) modulation of the neuroinflammatory environment, and (iii) enhancement of synaptic plasticity and tissue repair. Collectively, these actions support the structural and functional recovery of the AD-affected brain.
Conclusion
AD is a multifactorial disease with a complex pathophysiology. While there are many pharmacological treatments available, these drugs are limited to only improving symptoms and slowing disease progression and curative treatment for this disease is still lacking. Scientific information is constantly evolving, and in our article, we discuss the conventional treatment of AD and the present and future perspectives on the use of antidiabetics and stem cell therapy as promising therapeutic options in humans.

