











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Cutaneous leishmaniasis (CL) is the most common presentation of leishmaniasis, a disease caused by parasites of the Leishmania genus. American CL, also known as tegumentary leishmaniasis, is charachterized by skin ulcers throughout the body, resulting from the dissemination of parasites through the bloodstream and lymphatic system. The manifestation of symptoms depends on the individual’s immune response and the specific species of Leishmania involved (Pinart et al., 2020).
In America, CL is known as New World CL (NWCL) or tegumentary leishmaniasis, and it is mainly caused by L. mexicana and L. brasiliensis species complex (Abadías-Granado et al., 2021). In 2024, more than 34,000 new cases were registered in the region, with an increased incidence in Mexico, Argentina, Costa Rica, and Ecuador largely due to vector spread and population migration (PAHO, 2024).
Control and prevention are based on vector/reservoir control and pharmacological treatment, for which L. mexicana has shown intermediate to low sensitivity (Pinart et al., 2020). Treatment success is variable mainly due to parasites’ resistance, high costs, low availability in endemic areas and toxicity. Antimycotics like ketoconazole show some efficacy against L. mexicana but in vitro sensitivity studies have reported contradictory data (de Vries and Schallig, 2022).
Sometimes, the infection is self-resolved, which indicates an effective but incomplete immune response development, supporting that a vaccine could be used to treat or prevent leishmaniasis. Although several strategies have been used to achieve a vaccine for humans, there is no licensed candidate to date, thus hampering the efficient control of leishmaniasis. In the last decade, bioinformatics tools and genetic engineering have boosted leishmaniasis vaccines research (Dinc, 2022).
This review provides a comprehensive overview of current vaccine development against NWCL caused by L. mexicana, and examines remaining problems to achieving effective vaccines for humans. It also discusses the status of immunoinformatics applied to CL vaccine research, the challenges that remain to be addressed, and the perspectives of this approach applied to CL prevention and treatment.
New generation vaccines for nwcl
Live vaccination against CL, known as leishmanization (LZ), has been practiced for centuries in Middle East countries, but is not recommended due to safety issues. In addition, no evidence proves that LZ is protective against New World Leishmania species in humans (Moreira et al., 2023). However, LZ has shown that vaccination in endemic areas is the most cost-effective tool for leishmaniasis control and prevention. An ideal NWCL vaccine should fulfill some requirements: (i) good safety profile, (ii) minimum number of immunizations, (iii) cost-effectiveness, (iv) show prophylactic or therapeutic efficacy, (v) optimal delivery, and (vi) no need for cold chain supply (Rafati et al., 2017).
L. mexicana pathogenicity and the immune responses that mediate protection are complex. Nevertheless, a successful response to L. mexicana infection includes reactive oxygen species (ROS) and nitric oxide production by macrophages, which is triggered by T helper 1 lymphocytes, Natural Killer cells, and T cytotoxic lymphocytes. These immune responses decay faster than antibody responses (Abadías-Granado et al., 2021). Thus, immunological memory induction poses a problem for the effectiveness and efficacy of vaccines.
Several first (live or killed parasites), second (native or recombinant proteins) and third-generation (nucleic acids) vaccine candidates have been tested to prevent L. mexicana infection. Although there is still no vaccine for human use, advances reported in clinical trials provide hope for its development in the future (Moafi et al., 2019; Dinc, 2022) (Table I).
Tabla 1. Candidatos vacunales contra L. mexicana evaluados en modelos animales.
| Candidate | Antigens | Main results | Reference |
| DNA vaccine | L. mexicana GP63 gene into ΔaroD S. typhi strain CVD 908 | Protection against active CL in mice. Partial protection in monkeys. No need for adjuvant | (González et al., 1998) |
| (3rd generation) | |||
| Native proteins | Adjuvated GP63, CP, and MBA | Protection against promastigotes in C57B/L mice. Risk of transient and accentuated disease | (Aebischer et al., 2000) |
| (2nd generation) | |||
| L. mexicana H-line | Gentamicin-attenuated L. mexicana | Significant control of WT parasites through Th1 response in BALB/c mice | (Daneshvar et al., 2003) |
| (1st generation) | |||
| Killed parasites | Autoclaved L. mexicana + BCG | Effective as immunotherapy in human severe mucocutaneous and diffuse CL resistant to treatment | (Convit et al., 2004) |
| (1st generation) | |||
| Genetically modified L. mexicana | Δ GDP-MP (live attenuated) | Long-lasting protection in BALB/c mice. Risk of virulence reversion | (Zabala-Peñafiel et al., 2020) |
| (1st generation) | |||
| In silico predicted antigen | L. mexicana MBA gene into plasmid pVAX1 | Parasite reduction and improvement of clinical manifestations of CL in mice. No need for adjuvant | (Burgos-Reyes et al., 2021) |
| (3rd generation) | |||
| Genetically modified L. mexicana | LmexCen-/- | Induce immune response similar to natural infection. Risk of virulence reversion | (Volpedo et al., 2022) |
| (1st generation) |
GDP-MP: GDP-mannose pyrophosphorylase; lmlpg2: L. mexicana Golgi GDP-mannose transporter coding gene; CPB: Cysteine proteinase B; MBA: Membrane bound acid phosphatase; WT: wild-type.
Genome sequencing of L. mexicana species complex has enabled its genetic attenuation by targeted gene disruption (Saravia et al., 2006) and CRISPR/Cas9, with variable protection induction in murine and non-murine models (Volpedo et al., 2022). In contrast, Ishemgulova et al. (2018) demonstrated that L. mexicana knockout strains for a putative virulence factor predicted in silico, do not alter colonization in neither vector nor mice, which highlights the importance of bioinformatics-predicted virulence factors experimental validation.
Attenuated vaccines present drawbacks regarding standardization for large-scale production, virulence reversion or incomplete attenuation, and differences in protection between preclinical and clinical trials. Nevertheless, attenuation by genetic modification could contribute to maintain immunogenicity and vaccine potency, but assure no virulence reversion (Zabala-Peñafiel et al., 2020).
On the other hand, L. mexicana and L. amazonensis autoclaved promastigotes have been used as killed vaccines in South America, where cross-protection has been reported in some countries (Convit et al., 2004). Nevertheless, autoclaved parasites lose potency over time, therefore efforts are focused on combining its application with immunotherapy and chemotherapy (Zabala-Peñafiel et al., 2020).
Regarding second-generation candidates, subunit and recombinant vaccines for dogs have been authorized and are currently available in Europe and Brazil, namely Canileish®, Leish-Tec®, Leishmune® and Letifend® (Calzetta et al., 2020), supporting that a vaccine for humans is feasible. Although pathogen subunits do not confer long-lasting immunity, they are safer, more tolerated, and better characterized than whole-cell vaccines (Tahamtan et al., 2017).
Several attempts of second-generation candidates have been explored: fractionated parasite and vector proteins, polyprotein combinations, and delivery systems (Coler and Reed, 2005). However, Leishmania recombinant proteins are expensive to produce at large-scale, which is not viable for mass vaccination. Therefore, it is preferable to design synthetic polyepitopic vaccines using combinations of conserved epitopes. Moreover, synthetic peptides have great versatility to adapt to innovative delivery systems and have been investigated for vaccination against NWCL (Gupta et al., 2021).
Polyepitopic molecules have increased immunogenicity, lower risk of adverse reactions, and wider population/parasite species coverage compared to crude antigens. For instance, L. mexicana Nucleoside hydrolase 36 (NH36), a vital enzyme for parasite metabolism, has proven cross-protection against L. braziliensis in humans (Alves-Silva et al., 2019). Also, preclinical evidence indicates properly adjuvanted peptides and genetic vaccines induce strong protective cellular immunity (Graña et al., 2022).
In this regard, third-generation vaccines are advantageous due to their possibility to combine several epitopes into a single formulation that could provide cross-protection against different Leishmania species. NH36 and GP63 are the most tested proteins as genetic vaccines for NWCL in mice: VR1012-NH36 DNA candidate conferring cross-protection against L. chagasi and L. mexicana (Dumonteil et al., 2003), and naked gp63 DNA adjuvanted with aluminum induces cellular immune responses (Rosado-Vallado et al., 2005).
Several L. mexicana genes encoded in plasmid VR1012 have also been tested as DNA vaccines in mice challenges, inducing partial protection in all cases (Dumonteil et al., 2003). pVAX1 is another plasmid that has been used as a vector for an in silico predicted membrane-bound acid phosphatase gene from L. mexicana (LmMBA), demonstrating a protective effect in mice (Burgos-Reyes et al., 2021).
However, the failure of several genetic vaccine candidates demonstrates that protection against leishmaniasis is more complex than originally thought. Low immunogenicity of naked DNA vaccines’ due to degradation, hydrophilic nature, and poor recognition, is a major challenge (Akbari et al., 2021). Other concerns with nucleic acid vaccines are possible recombination with the host genome, enhanced disease and low gene transfection efficacy, which poses substantial problems for safety and manufacturing (Tejeda-Mansir et al., 2019).
Thus, researchers have explored alternative approaches including immunotherapy (Akbari et al., 2021), nanotechnology (Tejeda-Mansir et al., 2019), virus and bacteria (Cecílio et al., 2020). Remarkably, synthetic peptides, nucleic acids and proteins have great versatility to adapt to innovative delivery systems.
Cutaneous leishmaniasis vaccine development through reverse vaccinology
Reverse vaccinology (RV) involves the prediction of novel epitopes through bioinformatics, proteomics, comparative and functional genomics (Rappuoli, 2000). This methodology allows for thoughtful mapping and selection of immune targets with antigenic diversity, and has become extremely useful in vaccine research and development. Vaccine development against several pathogens and cancer has been swiftened using RV, thus holding great potential for global public health improvement (Cianci and Franza, 2022).
RV methodology and advances
Vaccine design through RV starts with retrieving genomic data (DNA or translation products) from public online databases that gather information and models for new vaccine targets and drug development. Despite gaps in L. mexicana metabolism and pathogenicity knowledge, there are some databases exclusively related to the parasite, such as LeishInDB, TriTrypDB, LeishPathNet, LeishDB, LeishCyc and LmSmdB. Using these resources, various RV strategies have been explored to design synthetic and chimeric peptides, as well as DNA vaccines (Flórez et al., 2021; Gupta et al., 2021). Figure 1 shows a general RV workflow.
Figure 1 Reverse vaccinology general workflow. In silico approach starts with antigen mining from genes/proteins and selection of vaccine candidate sequences. Next filtering levels involve immune epitopes prediction, selection and linking to merge a vaccine construction. Subsequently, secondary and 3D structure analysis is performed. Docking studies, and immunogenicity studies can also be conducted. Codon sequence optimization is also necessary prior to in vitro cloning. Vaccine candidate is further purified and validated before moving into clinical trials (Created under BioRender.com license).
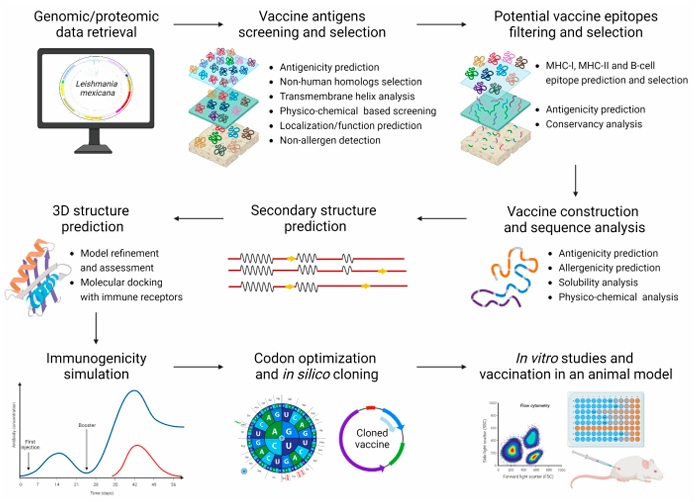
Figura 1. Flujo de trabajo general en vacunología reversa. El enfoque in silico comienza con la identifiación de antígenos a partir de genes/proteínas y con la selección de secuencias candidatas a vacunas. Los siguientes niveles de filtrado implican la predicción, selección y combinación de epítopos inmunogénicos en una construcción de vacuna. Posteriormente, se realiza un análisis de estructuras secundarias y se generan modelos tridimensionales de las proteínas. Además, se pueden realizar estudios de acoplamiento y estudios de inmunogenicidad. La optimización de la secuencia de codones es necesaria antes de la clonación in vitro. Por último, la vacuna candidata se purifica y se valida experimentalmente antes de pasar a los ensayos clínicos (Creado bajo licencia de BioRender.com).
L. mexicana genome comprises 8149 sequences on average, but gene expression varies depending on the parasite’s life stage (Rogers et al., 2011). Using DNA as a starting point for vaccine design offers the advantage of accessing all the potential proteins within the pathogen’s genome, but thousands of them are irrelevant as vaccine targets since they are not involved in cellular immune response (Calzetta et al., 2020). Considering this, an extensive analysis must be carried out to identify which disease-related genes are being expressed.
On the other hand, analyzing 3D structures of pathogenic proteins (translation products) offers valuable insights into the motifs responsible for immune recognition, thus helping in epitope prediction. Additionally, translation products can be obtained from diverse transcriptomic experiments, such as whole exome sequencing, RNAseq, or microarrays (Hwang et al., 2021). Using this approach, it is possible to assess the pathogen´s gene expression during multiple steps of its life cycle, and immune factors from the host, which leads to a more efficient antigen selection.
Numerous criteria for RV have been developed to aid in vaccine candidate prediction. The most widely used are the prediction of antigenicity, allergenicity and immunogenicity, subcellular localization, function, conservation, and physicochemical features such as hydrophobicity. However, for intracellular pathogens, subcellular localization could not be decisive, as antigens for T-cell immunity are not necessarily surface-exposed (Martinelli, 2022).
LmMBA, evaluated as a prophylactic vaccine by Burgos-Reyes et al. (2021) is a good example of a protein identified through a data mining approach. Various authors have developed immunoinformatics pipelines applicable to Leishmania vaccine design, with variable degrees of success when tested in vivo or in vitro (Singh et al., 2020; Rawal et al., 2021). This depends mostly on the parasite species, as well as target population, platforms/tools accuracy and procedure order, and the selected vaccine scaffold.
Once genomic data is retrieved, mining vaccine antigens is performed either by a subtractive genomic method or by a network-based approach. In the first case, the parasite transcriptome or proteome is classified according to each protein’s specific biological relevance while eliminating homologs that could lead to immune tolerance, autoimmune responses, or tissue damage in the host (Vivona et al., 2008).
In the network-based strategy, central proteins are identified in silico, a process that often involves mutational studies, molecular dynamics biology, and orthology-based methods to determine protein-protein interaction networks (Wheeler, 2021). The aforementioned methodologies can be combined, leading to an even deeper screening of potential targets. The selection process yields a short list of possible antigens to be tested in immunological simulations.
Conservation analysis among multiple pathogen strains and related species is also important. Such comparisons enhance our understanding of Leishmania diversity and permits identifying conserved epitope sequences specific to the pathogen, while minimizing or excluding variants found in other pathogen species that potentially elicit detrimental immune responses (Shams et al., 2022).
Cytokines like interferon gamma (IFN-γ) and major histocompatibility complex (MHC) I and II are important in protection against NWCL. One way to predict IFN-γ -inducing motifs is through IFNepitope, while MHC-II epitopes can be predicted in the Immune Epitope Database (IEDB), Vaxitop, and NetMHCIIPan-4.0, which is currently the most accurate software (Dhanda et al., 2013). Regarding MHC-I epitopes, artificial neural networks of NetMHC4.0 and NetMHCPan4.1 are reliable methods to screen for MHC-I binding peptides (Gonzalez-Galarza et al., 2020).
Genetic background can be detrimental for vaccine effectiveness. Since CL affects people in extensive areas, and MHC alleles are highly variable in humans, potential target population ethnicity must be taken into account. Population coverage, a tool from IEDB, in combination with allele frequency databases can be used to focus epitope prediction on a specific population based on geographical distribution. Herrera et al. (2020) created an interactive database with georeferenced information on Leishmania species distribution in America, which could help in target population selection and antigen/species conservation analysis.
B-cell epitopes may also be included in the vaccine to block the pathogen invasion. The corresponding analysis follows the same criteria of T-cell epitopes and can be performed alongside or after MHC selection. Tools like BepiPred3.0, ElliPro, DiscoTope and artificial intelligence platforms are available. Combining these softwares with a 3D visualization tool makes it possible to select linear and conformational B-cell epitopes (Woolums and Swiderski, 2021).
Regrettably, for L. mexicana no predicted immunogens have been successful in generating broadly neutralizing antibody responses. A highly specific and sensible B-cell epitope prediction for L. mexicana tool still does not exist because of epitope structural complexity. Another issue with epitope prediction from recombinant proteins is the loss of conformational epitopes as a result of non-native folding of the fragments (Martinelli, 2022). Therefore, not all predicted peptides are immunogenic in animal models.
Secondary structure analysis and 3D modeling of vaccine construction are critical. There are several tools for deducing chimeric protein structure: AlphaFold, RoseTTAFold and Modeller servers have become the most used for de novo predictions. However, depending on the amino acid sequence and program´s algorithm, the generated models could greatly differ between each other (Lee et al., 2022). Hence, it is recommended to compare the 3D models obtained from multiple tools and validate them using Ramachandran plots or servers like ERRAT, PROCHECK and ProSA-Web.
On the other hand, it is necessary to simulate the immune response induced by the vaccine, although sometimes the predictions do not match the results obtained in animal models (Rapin et al., 2010). Before preclinical evaluation, it is recommended to perform docking studies and immunogenicity simulations. PatchDock, AutoDock Vina, and SwissDock are the best performing docking simulators, while C-IMMSIM server is widely used for immunogenicity due to the variety of results it provides.
Finally, vaccines designed through RV may be used either as a subunit vaccine or as nucleic acid vaccine. Both approaches demand gene codon usage optimization, thus the peptide must be reverse translated into DNA and gene constructs adapted to improve cloning and expression. Servers like NovoPro and JCat are commonly used in this task, and gene synthesis companies often offer this service. It is also recommended to re-run the allergenic prediction test to prevent possible hypersensitivity reactions (Woolums and Swiderski, 2021).
RV challenges and future perspectives in NWCL vaccine research
RV methodology significantly reduces experimentation efforts and costs in comparison with the conventional approach. Furthermore, new-generation vaccines designed using in silico tools have proved to be safe, stable, and efficient in human and animal vaccination. The ultimate advantages of RV are speed, accuracy, and efficiency. These features contribute to cost-effectiveness of the vaccine development process, which is highly desirable in NTD research (Moxon et al., 2019).
However, in silico methods have drawbacks including failure in polysaccharide or glycolipid-derived antigens prediction, limited accuracy and reproducibility of antibody response simulation and immunogenic peptide ranking (Rappuoli, 2000). The RV approach is not inherently applicable to vaccine antigens that exhibit excessive variability, complex structures, or binding instability. In consequence, all predicted epitopes must be tested in vivo to determine their immunogenicity (Wheeler, 2021).
To date, RV´s main limitation is the lack of a high-throughput system to estimate memory immunity of selected candidates. Algorithms trained in a data set may not be able to make predictions in all proteomes or genomes, this fact is more evident when tools intended for one group of organisms are used to analyze information of an unrelated or distant species (Wheeler, 2021).
Although in silico methods present several drawbacks and shortcomings, their constant improvement through code development, AI-powered methods, and experimental validation has positioned bioinformatic methods as important tools for rational vaccine design. Molecular target discovery against L. mexicana is not a straightforward process, nevertheless bioinformatics tools combined with genetic engineering hold great promise due to their versatility.
New-generation vaccines in the reverse vaccinology era are promising strategies to design and develop vaccine candidates for human use. The foregoing approach has identified more potential vaccines against Leishmania than conventional approaches over the past 40 years. Considering this, it is not surprising that RV will be the method of choice for vaccinology studies in the near future.
Conclusions
RV has contributed to successfully identifying vaccine candidates from L. mexicana. However, several challenges need to be solved before achieving this task. An effective vaccine for L. mexicana must induce cellular immune responses, some of which require antigen persistence to be maintained, thus improved antigens and adjuvants should be investigated.
RV and immunoinformatics approaches allow vaccine design and evaluation in a relatively short time, although the need to invest in research, new diagnostic, treatment, and prevention strategies against CL remains. Finally, these efforts are expected to contribute towards the development of new generation vaccines for NWCL, tailored to both the genetic makeup of the human and the pathogen.

