











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
The strategy of identifying new therapeutic uses for existing drugs has long been a well-established approach in pharmaceutical research. However, recent advances in computational tools and omics technologies have brought renewed innovation to this field, enabling more systematic, large-scale, and data-driven repositioning efforts. These modern methodologies offer a cost-effective and time-efficient alternative to traditional drug development, by leveraging existing pharmacological and safety profiles to accelerate the discovery of new indications. This approach has proven particularly valuable in fields such as oncology (Xia et al., 2024), infectious diseases (Devillers, 2018; Pires et al., 2021; Pizzorno et al., 2019), and neurodegenerative disorders (Chaffey et al., 2022; Onódi et al., 2023). Despite its advantages, drug repositioning still faces challenges, including the identification of novel therapeutic targets and the experimental validation of computational predictions (Deshpande et al., 2020; Juárez‐Saldivar et al., 2021; Rashid et al., 2023). The objective of this review is to provide a comprehensive overview of drug repositioning as a strategy for accelerating drug discovery, with a particular focus on distinct family drugs. By integrating computational approaches, omics technologies, and experimental validation, this review aims to highlight current advances and in silico strategies to improve the identification and development of repositioned drug candidates.
Search strategy
A systematic literature search was conducted using the PubMed and ScienceDirect databases to identify relevant studies on drug repositioning. The search strategy involved the use of specific keywords. The primary search terms included “drug repositioning,” “drug repurposing,” and “therapeutic repositioning,” which were combined with different classes of drugs, such as “NSAIDs” and diseases, to ensure a comprehensive retrieval of relevant publications. To maintain the relevance and accuracy of the review, only articles published within the last fifteen years were considered, setting 2010 as the lower limit, prioritizing the most recent ones. This timeframe was chosen to reflect the most recent advancements in the field while ensuring that foundational studies were included. Studies were filtered based on relevance, prioritizing peer-reviewed articles, systematic reviews, and original research papers. Articles deemed outdated, redundant, or lacking methodological rigor were excluded.
Repurposing of non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) are a class of medications approved by the Food and Drug Administration (FDA) for their antipyretic, anti-inflammatory, and analgesic properties. They are commonly used to manage muscle pain, menstrual cramps, arthritis, fever, gout, and migraines, and as alternatives to opioids in some acute trauma situations (Sobhani et al., 2023). However, NSAIDs have demonstrated several activities than those originally reported, for example, acetylsalicylic acid (ASA), marketed and better known as Aspirin®, first was designed as an NSAID and then as an antiplatelet drug (Patrono and Rocca, 2021). The inhibitory action of this compound on blood platelets was not discovered until the late 1960’s, and it has been little more than a decade since this property was linked to the inhibition of the cyclooxygenase enzymes, mostly COX-1 through Ser530 prevents the production of prostaglandin and thromboxane A2, responsible for inducing platelet aggregation and vasoconstriction in endothelial cells. This mechanism of action explains the effect of ASA in preventing thrombosis of the cerebral vessels and coronary arteries (Fijałkowski et al., 2022).
In the last decade, new potential activities have been reported for ASA, including its antiviral properties. Its effect has been evaluated against a broad range of human pathogenic viruses, including adenovirus (Adeno 5), herpes simplex virus (HSV-1), human influenza A virus (FluA H1N1), respiratory syncytial virus (RSV), Coxsackie virus (CA9), and several human rhinoviruses (HRV). Specifically, ASA’s influence was analyzed in two HRV subtypes from both the “minor group” (HRV1A, HRV2) and the “major group” (HRV14, HRV39). In vitro studies were conducted using plaque reduction assays, demonstrating a dose-dependent effect of ASA against FluA H1N1, with a half-maximal effective concentration (EC50) value of 0.66 mmol/L. For CA9, HRV2, HRV14, and HRV39, the EC50 values were 0.98, 0.69, 0.21, and 0.1 mmol/L, respectively, whereas for the remaining viruses, the values exceeded 1 mmol/L. Interestingly, only RNA viruses were susceptible to the antiviral activity of ASA, suggesting a certain degree of specificity (Glatthaar-Saalmüller et al., 2017).
More recently, related to the specificity of previous work, ASA activity was tested against Bunyamwera virus (BUNV), a human pathogen RNA virus (Fernández-Sánchez et al., 2023). Authors use viral RNA polymerases as targets, proteins essential for viral genome replication and share sequence as well as structural similarities across different families of RNA viruses (Horst et al., 2019). Their approach was combining both ligand-based and structure-based molecular models, using a known influenza endonuclease inhibitor as reference 2,4-dioxo-4-phenylbutanoic acid (DPBA), obtaining FDA-approved drugs from DrugBank Database, and using crystal structure of the Hantaan virus endonuclease as target to obtain hit compounds through molecular docking. ASA was one of the hit compounds and was selected because of its similarity to DPBA. From the top five hit compounds, only ASA showed antiviral activity against BUNV, with an IC50 of 2.02 mM. ASA significantly reduced viral titters in BUNV-infected Vero cells, decreasing the expression of viral proteins Gc and N in a dose-dependent manner. ASA also inhibited the formation of Golgi-associated viral replication structures and protected the Golgi complex from BUNV-induced fragmentation, suggesting an interaction with vRNA polymerase that disrupts viral replication. Similar effects were observed with the vRNA polymerase inhibitor Ribavirin. Notably, the removal of ASA from the culture medium restored normal Golgi morphology and viral structures, indicating that its antiviral effects are reversible. While ASA effectively inhibits BUNV replication at 2 mM, its anticoagulant properties limit its use in hemorrhagic fever cases. However, ASA remains a cost-effective and safe candidate for broad-spectrum antiviral therapy, with potential applications against select bunyavirus infections (Fernández-Sánchez et al., 2023). The structural and functional similarities between influenza virus and bunyavirus endonucleases suggest that inhibitors designed for influenza could be repurposed to target bunyaviruses. This rationale guided an in silico screening of the DrugBank database, which includes approximately 10,000 compounds with prior human use. Using computational methods rather than manual selection allowed for a systematic evaluation of chemical similarities to DPBA, a known influenza endonuclease inhibitor. Virtual screening not only identified structurally related candidates, but also confirmed their compatibility with the catalytic site, a critical factor for effective inhibition. These findings underscore the importance of computational approaches in drug repurposing efforts, facilitating the identification of potential antiviral agents against bunyaviruses.
Several studies have explored the potential ASA benefits in COVID-19 patients, particularly regarding its antithrombotic and anti-inflammatory properties (Tantry et al., 2021). Observational cohort studies have reported that ASA use before or shortly after hospitalization was associated with reduced rates of mechanical ventilation, intensive care unit (ICU) admission, and in-hospital mortality, without significant differences in thrombosis or major bleeding (Chow et al., 2021). In American veterans with COVID-19, preexisting ASA prescriptions correlated with a lower mortality risk at both 14 and 30 days (Osborne et al., 2021). Similarly, in-hospital ASA use was linked to a decreased incidence of mortality in a propensity score-matched study (Meizlish et al., 2021). Another retrospective analysis suggested that ASA users had a lower risk of COVID-19 infection and a shorter disease duration (Merzon et al., 2021). Overall, prior and in-hospital ASA use at doses ranging from 75 to 325 mg per day has been associated with improved survival outcomes in COVID-19 patients (Liu et al., 2021).
Additionally, ASA has been studied to prevent cardiovascular diseases (CVD) linked to a first heart attack or stroke (Cofer et al., 2022). COX-1 and P2Y12 inhibitors are two classes of antiplatelet drugs approved to treat acute coronary syndrome and secondary prevention. Therefore, the combination of ASA (COX-1 inhibitor) and clopidogrel (P2Y12 inhibitor) is the standard treatment for atherosclerotic cardiovascular disease as per the 2015 European Society of Cardiology guidelines (Qaderdan et al., 2015; Saqallah et al., 2024).
ASA has presented activity on distinct types of cancer, like colorectal, where studies suggest that the reduction in risk of colorectal cancer is present in older people who have been using ASA since a younger age (Shureiqi, 2022). Recently, in silico and in vitro approaches showed that ASA also has good affinity to estrogen receptor alpha (ERα) and cause a decrease of mRNA levels of ERα followed by a decrease in protein expression, suggesting that ASA could be used as a potential anti-breast cancer agent (Guo et al., 2021; Kaur et al., 2024). In one study, ASA was docked onto three ERα crystals, each bound to a different type of ligand: agonist (1GWR), antagonist (5UFX), and selective estrogen receptor modulator (SERM) (3ERT) (Figure 1). The aim was to evaluate whether agonists (such as 17-β estradiol), antagonists (such as OP1074), or SERMs (such as tamoxifen) have unique binding patterns and to compare ASA’s behavior with them. The interactions between ASA and 3ERT showed HBs with residues Gly-521, Thr-347, and His-524, with His-524 being common in both antagonist-bound and SERM-bound ERα complexes. The Thr-347 interaction was exclusive to antagonist-bound ERα complexes. To assess the stability of ASA on different crystals, Molecular Dynamics (MD) studies were carried out. ASA was unstable on the 5UFX crystal, whereas ASA remained stable on the 1GWR and 3ERT crystals during a 500 ns simulation. The 3ERT + ASA complex showed a lower ligand RMSD (~1 Å), greater receptor stability, and higher average interaction energy (-26.86 kcal/mol) compared to the 1GWR complex (-22.73 kcal/mol), indicating stronger and more stable binding in the SERM-bound ERα conformation. Finally, following MD trajectories, authors found in 1GWR + ASA complex, ASA maintained HB with residues Arg-394, Glu-323, and Lys-449, and showed hydrophobic contacts with Pro-324. In 3ERT + ASA, it retained interactions with residues Gly-521, Thr-347, and His-524, as well as water bridge interactions with Ala-350 and Leu-525. These interactions were similar to those observed in antagonist- and SERM-bound ERα complexes, suggesting a potential antagonistic behavior of ASA. Secondary structure content, radius of gyration, and solvent-accessible surface area were comparable in both stable systems. Overall, 3ERT + ASA demonstrated more favorable dynamic and energetic properties, supporting its potential relevance in drug repositioning efforts, and his stability and interaction pattern suggests that ASA might behave like an antagonist (Kaur et al., 2024).
Figure 1 Crystallographic structures of estrogen receptor alpha (ERα) in complex with different types of inhibitors. A) ERα in complex with the agonist inhibitor 17-β estradiol. B) ERα in complex with the antagonist inhibitor OP1074. C) ERα in complex with the selective modulator inhibitor tamoxifen. Each ERα chain is represented in a different color (yellow, blue, green, and purple), and the inhibitor is shown in red. The structure of the inhibitor is displayed below each crystal.
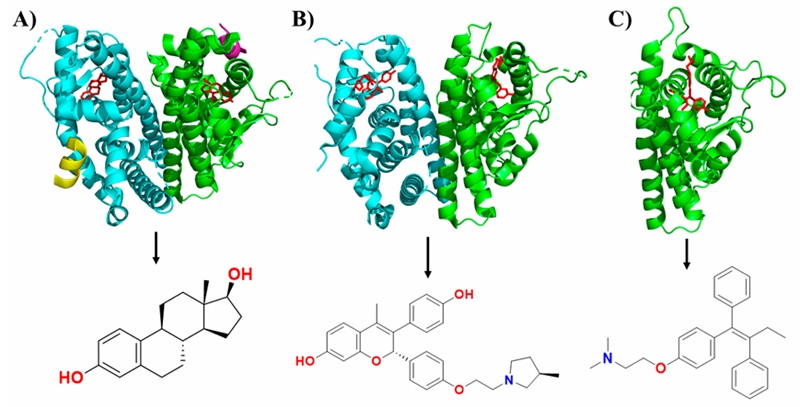
Figura 1. Estructuras cristalográficas del receptor de estrógenos alfa (ERα) en complejo con diferentes tipos de inhibidores. A) ERα en complejo con el inhibidor agonista 17-β estradiol. B) ERα en complejo con el inhibidor antagonista OP1074. C) ERα en complejo con el inhibidor modulador selectivo tamoxifen. Cada cadena del ERα se representa en un color diferente (amarillo, azul, verde y púrpura) y el inhibidor se muestra en color rojo. La estructura del inhibidor se muestra en la parte inferior de cada cristal.
Another example of NSAID repurposing is ibuprofen, a drug developed as an anti-rheumatic in the 1960’s, that now is a drug with anti-inflammation and antipyretic properties, and it’s one of the analgesics mostly used worldwide. A few studies have also explored NSAID-based treatments targeting key features of cystic fibrosis (CF) lung disease, particularly chronic infections and inflammation, which are major contributors to morbidity in CF patients (Lands and Stanojevic, 2016). Ibuprofen not only apports to the treatment of CF because of its anti-inflammatory activity but also has antibacterial and antifungal activities that could combat the causes of the inflammation. For example, Shah et al. (2018) tested the activity of ibuprofen against CF-associated Gram-negative pathogens. Their results showed that ibuprofen reduces in vitro the growth of different Pseudomonas aeruginosa strains (PAO1, M57-15, T63547, and H25815) and Burkholderia spp. in a dose-dependent manner. P. aeruginosa had a significant reduction in ATP after 100 μg/mL, 30-min exposure to ibuprofen. In vivo studies achieved lower CFU counts of P. aeruginosa PAO1 in the lung and the spleen, and a significant survival advantage (92%) was observed in mice after ibuprofen oral treatment (Shah et al., 2018).
Antiviral activity has also been studied for ibuprofen. From a dataset that contained 382 experimental and 267 approved drugs, Veljkovic et al. (2015) carried out a blind molecular docking on glycoprotein GP1 from the Ebola virus. This protein is not crystalized, but they previously had obtained a model (Veljkovic et al., 2015). Results suggested that ibuprofen binds to Elastin Microfibril Interface Located Proteins (EMILINs) binding domain in GP1, involved in interaction between GP1 and endothelial extracellular matrix (ECM) (Veljkovic et al., 2015). This is just an in silico approach but it was a good start for further investigation of ibuprofen against the Ebola virus.
Recently, Borba-Santos et al. (2021) found that ibuprofen had low in vitro antifungal activity against Sporothrix brasiliensis and S. schenckii (MIC median of 256 μg/mL), but when is co-administrated with antifungal drugs (amphotericin B, itraconazole, and terbinafine), it was able to reduce the concentrations required to inhibit fungal growth in vitro. In this study, the best combination obtained was ibuprofen plus amphotericin B against S. schenckii. This combination achieved a great decrease in the dose of amphotericin B (up to 125-fold) (Borba-Santos et al., 2021). Given the high toxicity of amphotericin B and the associated side effects experienced by patients, as well as the possibility that these effects are replicated in vivo, reducing the dose of this drug could represent a significant benefit for the treatment of the patient due to their joint activity with ibuprofen.
Repurposing of sildenafil
Drugs repurposed through serendipity are also an example of success. Sildenafil, a phosphodiesterase-5 (PDE5) inhibitor (Figure 2) also known as Viagra®, started clinical development as an agent for the treatment of hypertension and angina, a disease that is characterized by present chest pain caused by the less quantity of blood that arrives at the heart, and subsequently evolved into a revolutionary new oral treatment for erectile dysfunction and pulmonary hypertension during clinical trials (Ghofrani et al., 2006).
Figure 2 Crystallographic structure of phosphodiesterase 4 (PDE4) with sildenafil bounded to the catalytic pocket. The three-dimensional structure of PDE4 is shown in grey. A sulfate ion is represented in red-yellow, magnesium in green, zinc in black, and sildenafil in blue. The chemical structure of sildenafil is shown on the right.
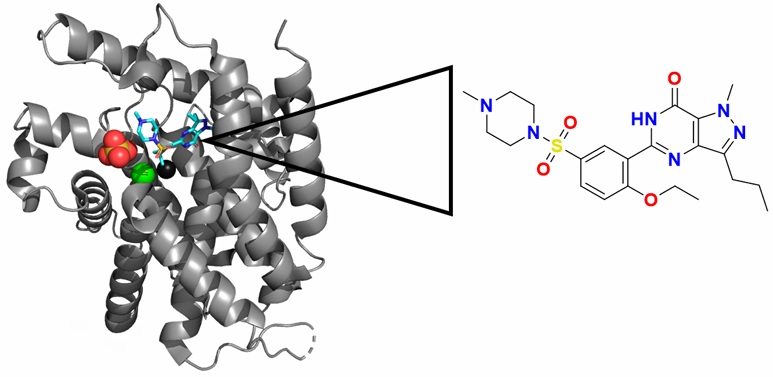
Figura 2. Estructura cristalográfica de la fosfodiesterasa 4 (PDE4) con sildenafil unido a la cavidad catalítica. En color gris, la estructura tridimensional de PDE. En rojo-amarillo, un sulfato. En verde, magnesio. En negro, zinc. En azul, sildenafil. A la derecha se muestra la estructura del sildenafil.
Nitric oxide (NO) functions as an inhibitory neurotransmitter in the gastrointestinal system, promoting the production of guanosine 3′,5′-cyclic monophosphate (cGMP) following swallowing. This process reduces lower esophageal sphincter (LES) pressure by inducing smooth muscle relaxation. The cGMP that accumulates is eventually broken down by PDE-5. Sildenafil, a powerful PDE-5 inhibitor, helps maintain cGMP levels by preventing its degradation, which leads to continued smooth muscle relaxation. Although, sildenafil is primarily used to treat erectile dysfunction by relaxing smooth muscle in the arterioles of the corpus cavernosum, it also reduces LES pressure, decreases esophageal propulsion, and slows gastric emptying (Wong et al., 2021).
In the last decade, sildenafil has also demonstrated to improve the antineoplastic drugs effects, for example, Das et al. (2010) obtained an increased antineoplastic activity when they combined sildenafil with doxorubicin (a broad-spectrum antitumor-antibiotic that has been widely used for treatment of several cancers, including breast, ovarian, and prostate cancers) against bone metastasis PC-3 and human prostate cancer DU145 cells. Doxorubicin alone induces apoptosis of 7.52% and 45.02% of PC-3 and DU145 cells, respectively. Co-treatment with sildenafil and doxorubicin increases apoptosis in both cell lines (18.71% in PC-3 and 56.82% in DU145 cells). Interestingly, sildenafil doesn’t have an effect alone against these cell lines (Das et al., 2010). In 2020, another study showed that exposure of the breast cancer cell line MDA-MB-231 to low concentrations of sildenafil (25 µM) inhibited cancer cell proliferation, promoted apoptosis, and reduced tumor growth. This antitumor effect was linked to sildenafil’s impact on the expression of HSP90, a molecular chaperone that facilitates the degradation of protein kinase D2 (PKD2), a serine/threonine kinase crucial for maintaining cancer cell proliferation and viability (Chen et al., 2020).
Many cancers are characterized by PDE5 overexpression, which suggests their involvement in carcinogenesis, and may be linked to the variation in the ratio of cGMP to cAMP and PDE levels among healthy and malignant tissues, notably in those with higher cGMP PDE activity (Savai et al., 2010). Therefore, sildenafil was evaluated in vivo and in silico as antineoplastic potential against Ehrlich Ascites Carcinoma. Sildenafil, both as a mono-treatment and in combination with cisplatin, significantly reduced tumor cell count, viability, growth rate, and proliferation while promoting apoptosis and causing cell cycle arrest at G0/G1 and sub-G1 phases. It also enhanced the cellular immune response by increasing plasma levels of granzyme B and IFN-γ, and improved the proportion of cytotoxic (CD3+) and helper T cells (CD3+CD4+) in the spleen while decreasing regulatory T cells. To evaluate the potential mechanism of action of sildenafil, some crystals as Major Histocompatibility (MHC), Lymphocyte-Specific Protein Tyrosine Kinase (Lck), Mitogen-Activated Protein Kinases (MAPKs) and PDE, were retrieved to molecular docking. Sildenafil achieved excellent docking scores -17.5 and -18.1 Kcal/mol with Lck and MAPKs, respectively, very close to the docking score achieved by sildenafil with PDE (-18.3 Kcal/Mol). Docking of sildenafil with MHC achieved only a docking score of - 6.4 Kcal/mol. To this end, LcK and MAPKs were identified as potential targets for sildenafil, and their docking results were further analyzed. sildenafil was found to bind in the ATP binding site of Lck, with a docking score of -17.5 kcal/mol, forming a HB with the crucial gatekeeper residue Thr316. Additionally, sildenafil interacts with Asp382, Gly252, and Gly254 through a HB and with Lys273 via an arene interaction. In contrast, the co-crystal ligand binds with Val259 through an HB and with Thr316 and Met319 through HBs. On the other hand, sildenafil exhibited a similar binding pattern to the co-crystallized ligand in the p38α MAPK binding site, with docking scores of -18.3 and -17.9 kcal/mol, respectively. The nitrogen atom of sildenafil’s piperidine ring forms a HB with Lys53 in the active site, while the co-crystal ligand interacts with Lys53 via one HB and two arene interactions. sildenafil also forms hydrophobic bonds with Met109 in the hinge region, while the co-crystal ligand accepts HB. Additionally, sildenafil demonstrated enhanced binding through three hydrophobic interactions with Gly31 and Val30. In conclusion, in silico data suggest that Lck and MAPKs are potential targets of immunomodulatory, apoptotic, and anti-proliferative sildenafil’s activity (Morsi et al., 2023).
In a recent study, sildenafil showed a reduction in the likelihood of Alzheimer’s Disease (AD), with a 54% reduced incidence, finding that sildenafil treatment reduced taurine hyperphosphorylation (pTau181 and pTau205) in a dose-dependent manner in both familial and sporadic AD patient isogenic pairs of wild-type and mutant induced pluripotent cell (iPSC) derived neurons. Moreover, the same study showed that sildenafil targets AD-related genes and pathobiological pathways, supporting the potential of this erectile dysfunction drug as a candidate drug for the treatment of AD (Gohel et al., 2024).
The expanding therapeutic potential of sildenafil opens new avenues for both neurological and oncological treatments. Its capacity to reduce tau hyperphosphorylation and modulate Alzheimer’s disease-related pathways suggests that it could become a viable option in the development of disease-modifying therapies for neurodegenerative disorders not only tested alone but also tested as a co-treatment with other reported compounds of these diseases (Gohel et al., 2024).
Fang et al. (2021) explored the potential of repurposing sildenafil as a treatment for AD using an endophenotype-based in silico network. Sildenafil was significantly associated with a 69% reduced risk of AD, based on retrospective pharmaco-epidemiological analyses of insurance claims data from 7.23 million individuals. This association remained consistent across multiple drug cohorts, even after adjusting for factors such as age, sex, race, and comorbidities. Mechanistically, sildenafil was shown to increase neurite growth and decrease phospho-tau expression in neuron models derived from AD patient-induced pluripotent stem cells, suggesting a potential therapeutic effect for AD. Furthermore, the study demonstrated a network-based approach for AD drug discovery, integrating multi-omics data from AD transgenic mouse models, which improved predictions for potential treatments. Comprehensive subgroup analyses also indicated that sildenafil was associated with a reduced risk of AD across various demographic and health condition subgroups, strengthening its potential as a candidate for AD prevention or treatment. Overall, these findings highlight sildenafil’s promise as a repurposed drug for AD, supported by both epidemiological data and mechanistic evidence (Fang et al., 2021).
Ongoing repurposing of proton-pump inhibitors
Proton-pump inhibitors (PPI) are a group of benzimidazole-substituted compounds that are employed to treat gastric and duodenal ulcers, gastroesophageal reflux disease, and other excessive acid-related secretory disorders. PPIs as omeprazole and their analogues (esomeprazole, pantoprazole, lansoprazole and rabeprazole) since 2011 have been studied as new antiprotozoal agents.
Pérez-Villanueva et al. (2011) reported PPIs antiprotozoal activity against Trichomonas vaginalis, Giardia intestinalis, and Entamoeba histolytica. All PPIs had activity in the nanomolar range against the three parasites. PPIs were 1.9-3.1 times more active than metronidazole against T. vaginalis and 12.8-78.1 times more active against G. intestinalis. Besides, rabeprazole and pantoprazole were 14.8 and 134.6 times more active than metronidazole against E. histolytica. The authors highlight the activity of pantoprazole against the three anaerobic protozoa tested (with IC50 values of 0.0756, 0.0157, and 0.0026 µM against T. vaginalis, G. intestinalis, and E. histolytica, respectively) (Pérez-Villanueva et al., 2011).
After, Reyes-Vivas et al. (2014) showed the mechanism of action of PPIs, specifically omeprazole, on triosephosphate isomerase of G. lamblia (GlTIM). This activity is linked to the inactivation of triose phosphate isomerase (TIM) of the parasite in a specific manner through interaction with Cys 222 (Figure 3), because PPIs bind covalently to sulfhydryl groups of cysteine residues deriving in the cytotoxic effect caused by omeprazole on the trophozoites of G. lamblia, proposing GlTIM as a potential target for drug design (Reyes-Vivas et al., 2014).
Figure 3 Crystal structure of Giardia intestinalis triosephosphate isomerase in its homodimeric form (PDB: 4BI7) and proton pump inhibitors structure. Since only mutant crystals of this protein are available with altered cysteines, the structure was modeled to represent the wild-type form (only the Ala202Cys mutation was introduced). A) Cysteines 14, 127, 202, and 228 are shown in yellow in stick representation. Cysteine 222 is shown in pink; this residue is derivatized by proton pump inhibitors, leading to its giardicidal activity. B) Structure of proton pump inhibitors. From top to bottom: omeprazole, esomeprazole, pantoprazole, lansoprazole, and rabeprazole.
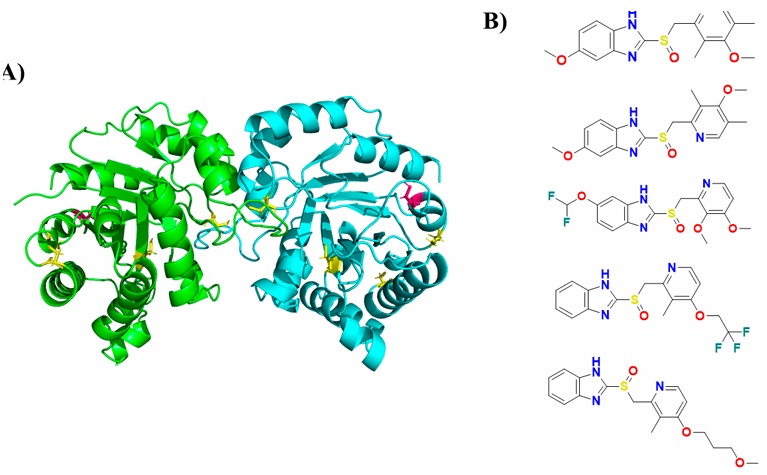
Figura 3. Estructura cristalográfica de la triosa fosfato isomerasa de Giardia intestinalis en su forma homodimérica (PDB:4BI7) y estructura de los inhibidores de la bomba de protones. Dado que existen solo cristales mutados en las cisteínas de esta proteína, se modeló para tenerlo en su forma Wild-type (únicamente se hizo el cambio Ala202Cys). En color amarillo, las cisteínas en representación stick (cisteínas 14, 127, 202 y 228). En color rosa, la císteina 222, la cual se derivatiza por los inhibidores de la bomba de protones para causar su actividad giardicida. B) Estructura de los inhibidores de la bomba de protones. De arriba abajo: omeprazol, esomeprazol, pantoprazol, lansoprazol y rabeprazol.
García-Torres et al. (2016) tested the effects of PPIs on the activity of GlTIM wild-type. The exposure of this glycolytic enzyme to esomeprazole, pantoprazole, and lansoprazole induced the inactivation of GlTIM in a very similar manner to omeprazole, losing 100% of its activity when these PPIs were incubated at 1 mM. Prominently, the efficacy of rabeprazole is remarkable because the concentration required to completely inactivate GlTIM was only 0.12 mM. The IC50 values of PPIs were approximately 0.30 mM, while the IC50 of rabeprazole was close to 0.03 mM. Furthermore, human TIM (HsTIM) was resistant to all concentrations of PPIs tested in this study, highlighting that when 50% of GlTIM was lost, HsTIM remained at almost 100% of its original activity (García-Torres et al., 2016). This study provides valuable insights into how structurally similar compounds, like omeprazole and its analogs, could potentially be repurposed for targeting conserved proteins such as TIM (target of interest for protozoan infections). While evidence of the activity of PPIs against various protozoa has been observed, further research is necessary. Specifically, modifications to the formulation or pharmaceutical presentation of PPIs may be required to enhance their efficacy against protozoan parasites. The widespread use and approval of PPIs for treating acid-related disorders could streamline the development process for therapies targeting protozoan infections, saving both time and resources in the development of new treatments.
Ongoing repurposing of antidiabetics
Antidiabetics are part of a large group of drugs that are directed to reduce polysaccharide degradation and intestinal glucose absorption, decrease hepatic glucose production, or increase insulin secretion directly and indirectly (T. A. Deshpande et al., 2022). Interestingly, this family has been well studied on different biological activities such as antibacterial (Zhen et al., 2024), antiparasitic (Juárez-Saldivar et al., 2020; Rub et al., 2019; Vázquez et al., 2024), AD (Shi et al., 2023), among others.
Ipragliflozin is an oral antidiabetic drug that belongs to the class of S sodium-glucose co-transporter 2 (SGLT2) inhibitors. It works by inhibiting the SGLT2 protein in the proximal tubule of the kidney, which reduces glucose reabsorption and increases glucose excretion through the urine. Its primary indication is the treatment of type 2 diabetes mellitus (T2D) (Alkabbani and Gamble, 2021). Zhen et al. (2024) did in silico and in vitro approaches where ipragliflozin presented anti-Mycobacterium tuberculosis activity. In this study, they predicted Rv1151c (a bifunctional enzyme with both deacetylation and desuccinylation activities, which plays an important role in M. tuberculosis drug resistance and stress responses) crystal structure and identified binding sites based on the druggability score. Once they chose what they called binding site 1, a structure-based virtual screening against predicted Rv1151c structure was carried out in site 1. Ipragliflozin is predicted to bind deeply within binding pocket 1 of the Rv1151c protein. Several residues-including Tyr183, Pro184, Leu155, Phe151, and Trp150-contribute to hydrophobic interactions with the ligand. Additionally, ipragliflozin forms one HB with each of the residues Ser179 and Thr178, two HBs with Gln86, as well as two π-π interactions with Phe151 and one π-π interaction with Phe20. MD study showed ipragliflozin and Rv1151c are stable after 55 ns. Finally, in vitro assay against M. tuberculosis H37Rv was made, demonstrating ipragliflozin MIC90 of 0.1 mg/mL in a dose-dependent manner, indicating that ipragliflozin is an anti-M. tuberculosis agent by targeting Rv1151c predicted structure (Zhen et al., 2024).
Glyburide, gliquidone, and glipizide, part of the sulfonylureas family (second-generation), were studied against the parasite Trypanosoma cruzi, the ethiological agent of Chagas disease (Gómez-Ochoa et al., 2022). Juárez-Saldivar et al. (2020) carried out a study with an in silico and in vitro approach of FDA-approved drugs against Chagas disease. Ten potential compounds where selected through molecular docking of a 1857 FDA-approved drugs library on the bifunctional enzyme dihydrofolate reductase-thymidylate synthase (DHFR-TS) of T. cruzi, and tested on epimastigotes, the cultivable form of the parasite. Glypzide showed interactions with amino acids Ala28, Ser83, and Gly156 through HB and forms hydrophobic interactions with Ile41, Phe52, Phe88, and Leu91, Glyburide forms HB with Ala28, Ser83, Ile154, Gly156, and Tyr160, while also interacting hydrophobically with Ile35, Ile41, and Thr80, and Gliquidone interacts with Ala28, Gly156, and Tyr160 through HB, and forms hydrophobic interactions with Ile35 And Thr80, along with π-stacking with Phe52. They found that glipizide, glyburide, and gliquidone presented the lowest free energy of binding against DHFR-TS, and had growth inhibitory activity, with IC50 values of 6, 13.4, 12, and 66 µM, respectively. Authors reported that residues Ile-41 and Phe-52, corresponded to the interaction profile of known DHFR-TS inhibitors (Figure 4), which are present in the three antidiabetics drugs mentioned (Juárez-Saldivar et al., 2020)
Figure 4 Three-dimensional structure of Trypanosoma cruzi dihydrofolate reductase-thymidylate synthase and antidiabetic compounds with trypanocidal activity. A) Three-dimensional structure of dihydrofolate reductase-thymidylate synthase. In red, isoleucine 41 and phenylalanine 52 amino acid residues, identified by the authors as important for determining whether a compound has inhibitory potential against the enzyme. B) Antidiabetic compounds that showed activity against the enzyme. From top to bottom: glyburide, gliquidone, and glipizide.
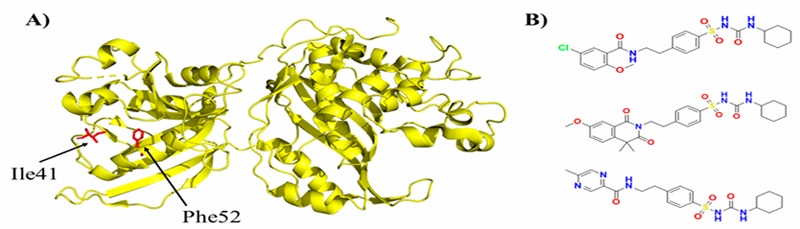
Figura 4. Estructura tridimensional del dihidrofolato reductasa-timidilato sintasa de Trypanosoma cruzi y los antidiabéticos con actividad tripanocida. A) Estructura tridimensional de dihidrofolato reductasa-timidilato sintasa. En rojo, los residuos isoleucina 41 y fenilalanina 52, señalados por los autores como importantes para determinar si un compuesto presenta potencial de inhibición en la enzima. B) Antidiabéticos que presentaron actividad contra la enzima. De arriba abajo: gliburida, gliquidona y glipzida.
More recently, these three sulfonylurea drugs have been tested in combination with benznidazole (Bzn) against T. cruzi. This study showed that in monotherapy, the antidiabetic drugs demonstrated antiparasitic effects by preventing the progression of infection (release of trypomastigotes), with an IC50 ranging from 8.4 to 14.3 µM, compared to Bzn, which had an IC50 of 0.26 µM. However, in combination therapy, the addition of just 0.5 or 1 µM of the antidiabetics reduced the IC50 of Bnz by 5 to 10 times, bringing it down to 0.03-0.05 µM. Notably, when used alone, the antidiabetic drugs reduced the infection in mice by 40-60%, similar to the 80% reduction observed with Bnz. Furthermore, the combination of Bnz and antidiabetics altered the antioxidant metabolites in epimastigotes. Glyburide and gliquidone alone decreased the levels of glutathione and trypanothione and increased those of cysteine, while glipizide alone increased the level of trypanothione and decreased the levels of cysteine and glutathione. The changes in these antioxidant metabolites suggested perturbation of the antioxidant system in the parasite (Vázquez et al., 2024). This research highlights the importance of continuing the investigation of previously reported drugs with activity, as well as the consideration that, as observed in the parasite’s antioxidant system, potential repurposed drugs may have multiple targets. Therefore, enzymes of the T. cruzi antioxidant system, such as glutathione synthetase, trypanothione synthetase, and trypanothione reductase, could be evaluated in conjunction with DHFR-TS.
Also, glyburide showed activity against the parasite Leishmania donovani using an in vitro and in silico approach. Molecular docking was conducted against Leishmania donovani-Trypanothione synthetase (LdTrySyn), a critical enzyme for the parasite’s survival as it detoxifies nitric oxide species produced by the host’s immune system. Results showed that glyburide interacts effectively with key residues in the LdTrySyn’s catalytic site as Asp348, Ser349, and Arg553, and in vitro growth reversibility assay shows that growth of treated parasite was not reversed when transferred to fresh culture media after 7 days, highlighting LdTrySyn as a drug target against L. donovani (Rub et al., 2019).
Metformin, derived from a plant named Galena officinalis, is currently widely used as a first-line drug for the treatment of T2D. Some studies have proved the effect of metformin on the prophylaxis and treatment of neuropsychiatric degenerative diseases in addition to its significant hypoglycemic effects, especially in patients with T2D (Orkaby et al., 2017). Shi et al. proposed an in silico approach to establish a relationship between T2D and AD. From 89 targets, catalase presented the best free binding energy with metformin. Metformin was predicted to interact with Glu330 and Ser120 twice. The cat-metformin complex was selected for 50 ns MD simulation and system stability analysis. The CAT-metformin complex was relatively stable at around 30 ns, the RMSD fluctuates at 0.2 nm (2 Å), and the stability was given by Asp259 amino acid residue through three HBs (Shi et al., 2023). However, these results are just based on computer simulations and big data analysis, which need to be further verified by cell or animal experiments.
Conclusions
In this review, case studies of NSAIDs, such as ASA and ibuprofen, illustrate the potential of repositioning well-known drugs for treating diseases beyond their original indications. For example, ASA has demonstrated antiviral properties, while ibuprofen has shown promise in treating chronic infections, and inflammation in cystic fibrosis. These findings emphasize the importance of combining computational, experimental, and clinical approaches in the repositioning process, offering valuable insights into the broader therapeutic potential of these drugs.
Similarly, sildenafil’s unexpected shift from a hypertension treatment to a successful therapeutic option for erectile dysfunction and pulmonary hypertension exemplifies how serendipitous discoveries can significantly impact drug repurposing strategies. This further illustrates the vast untapped potential within the pharmacological profiles of already approved drugs.
The ongoing repurposing of PPIs and antidiabetic drugs highlights their promising potential as therapeutic agents against parasitic infections. PPIs, traditionally used to treat acid-related disorders, have shown significant antiprotozoal activity, particularly against Trichomonas vaginalis, Giardia intestinalis, and Entamoeba histolytica. The observed efficacy of these compounds, notably pantoprazole, against various anaerobic protozoa, along with their ability to specifically target parasite enzymes like triose phosphate isomerase (TIM), positions them as valuable candidates for further investigation in parasitic drug development. However, optimizing their formulation and pharmaceutical presentation remains a crucial step in enhancing their therapeutic potential.
Similarly, antidiabetic drugs such as ipragliflozin, sulfonylureas (glipizide, glyburide, and gliquidone), and metformin have demonstrated significant antiparasitic effects. Ipragliflozin’s activity against Mycobacterium tuberculosis, along with sulfonylureas’ ability to inhibit key enzymes in T. cruzi and L. donovani, underscores the broad-spectrum potential of these drugs. Notably, the combination of these drugs with established therapies like Bzn has been shown to improve treatment outcomes, indicating the value of repurposing these agents. Furthermore, metformin’s potential in treating neurodegenerative diseases such as AD adds another layer to its therapeutic utility, particularly in the context of its established role in managing type 2 diabetes.
The concept of drug repositioning has evolved significantly with the integration of advanced computational tools and omics technologies, making it a valuable approach for discovering new therapeutic uses for existing drugs. By leveraging the extensive safety and pharmacological profiles of approved drugs, repositioning provides a faster and more cost-effective alternative to traditional drug development. This strategy has shown considerable promise in various therapeutic areas, including oncology, infectious diseases, and neurodegenerative disorders. However, challenges remain, particularly in identifying novel therapeutic targets and validating computational predictions through experimental methods.

