nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
SARS-CoV-2 genome analysis has revealed between 79.6 % to 80 % of genetic identity with SARS-CoV, showing similarities in proteins encoding sequences and viral infection mechanism (Andersen et al., 2020; Shereen et al., 2020; Wu et al., 2020; Zhou et al., 2020). Genomic study showed a close phylogenetic relationships between SARS-CoV-2 and SARS-CoV genomes, suggesting a similar evolutionary origin, however, SARS-CoV-2 has better host adaptation (Chen et al., 2021).
Keys proteins for a successful replication have been reported in both viruses, the nucleocapsid protein (N) and ORF6 protein (Kopecky-Bromberg et al., 2007; Gao et al., 2021). Nucleocapsid protein plays crucial roles such as viral genome protection and traffic to exocytic vesicles, but the crucial function is blocking interferon-mediated antiviral response (Shereen et al., 2020; Li et al., 2020; Iqbal et al., 2020; Kannan et al., 2020; Kopecky-Bromberg et al., 2007; Qinfen et al., 2004; Rowland et al., 2005; Timani et al., 2005; Surjit et al., 2004). A recent study suggests as a possible subcellular localization for SARS-CoV-2 N protein, the nucleus of the host cell (Gao et al., 2021). The presence of N proteins in the nucleus/nucleolus of infected cells has been described in SARS-CoV infection, due to structural exposure of nuclear localization sequences (NLS) in regions of the C-terminal domain, which is recognized by importins α (Timani et al., 2005; He et al., 2004; Wulan et al., 2015; Wurm et al., 2001).
ORF6 protein in coronaviruses is intimately related to importins α functions, specifically to importin α1, but the presence of this protein inside the host nucleus has not been evidenced yet (Gordon et al., 2020; Frieman et al., 2007; Hussain and Gallagher, 2010; Zhao et al., 2009). ORF6 and N proteins are essential for the inhibition of the interferon signaling pathway during infection with both viruses (SARS-CoV and SARS-CoV-2) (Frieman et al., 2007; Li et al., 2020; Liu et al., 2014; Ye et al., 2008; Zhao et al., 2009).
Importins α are responsible to NLS recognition in proteins that will be transported into the nucleus, playing an important role mediating antiviral responses through STAT1/STAT2 pathway (Pumroy and Cingolani, 2015). Several studies have shown that viruses are able to use this cellular transport pathway, specifically to import viral proteins into the nucleus and to block antiviral response mechanism of infected cells (Gayozo and Rojas, 2021; Pumroy and Cingolani, 2015).
An in vitro study has shown that ivermectin is able to decrease the SARS-CoV-2 viral genome load about 5000 fold in 48 hours post-treatment, the researchers suggest via blockage in the trafficking of viral proteins between nucleoplasm and cytoplasm (Caly et al., 2020). One of the key mechanisms of action reported for ivermectin molecules against SARS-CoV-2 infection is the binding affinity to importin α/β1 heterodimers, as well as for other karyopherins (KPNA/KPNB) receptors, inhibiting viral protein transport into the nucleus of the infected cell, however, it is still unknown how protein transport inhibition acts against SARS-CoV-2 proteins translocation (Zaidi and Dehgani-Mobaraki, 2022).
Due to this, protein-protein and protein-ligand molecular docking simulation were performed between ivermectin, N and ORF6 proteins of SARS-CoV-2, to human importins α isoforms as main target. In addition to this, active residues, molecular interactions in complexes were identified to understand the interactions modes between ivermectin, SARS-CoV-2 N and ORF6 proteins to importins α isoforms encoded in human genome, and to recognize possible molecular targets for the development of new antivirals against COVID-19.
Material and methods
Analysis of similarities between N and ORF6 proteins sequences
Nucleocapsid amino acid sequences of SARS-CoV-2, SARS-CoV, MERS-CoV, HCoV-OC43, HCoV-NL63, HCoV-HKU1, HCoV-229E and the ORF6 protein sequences of SARS-CoV-2 and SARS-CoV, belonging to Coronaviridae family, were obtained from the NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/) database (Table 1). These sequences were selected taking into account the infectious capacity of the most common coronaviruses that affect humans and the degree of conservation of the sequences, following the methodology proposed by Ibrahim et al. (2020).
Tabla 1. Código de accesión de las secuencias de las proteínas N y ORF6.
| Virus | Accession code | Database |
| Nucleocapsid protein | ||
| SARS-CoV-2 | QHO62110.1 | NCBI (Protein) |
| SARS-CoV | ABA02277.1 | NCBI (Protein) |
| MERS-CoV | AZK15907.1 | NCBI (Protein) |
| HCoV-OC43 | QBP84763.1 | NCBI (Protein) |
| HCoV-NL63 | ABE97134.1 | NCBI (Protein) |
| HCoV-HKU1 | ARU07581.1 | NCBI (Protein) |
| HCoV-229E | ARU07605.1 | NCBI (Protein) |
| ORF6 Protein | ||
| SARS-CoV-2 | QIV65092.1 | NCBI (Protein) |
| SARS-CoV | NP_828856.1 | NCBI (Protein) |
Relationships between proteins sequences were determined by phylogenetic reconstruction, using neighbor joining method with a bootstrap of 1000 replicates and Poisson substitution model using MEGA X software (Kumar et al., 2018). Alignment between amino acid sequences were performed using Clustal Omega algorithm (Sievers and Higgins, 2014), and BLOSUM 62 matrix between SARS-CoV-2 and SARS-CoV sequences to determine identity and similarity between them and record the conserved regions, for it EMBL-EBI tools (https://www.ebi.ac.uk/jdispatcher/psa) (Li et al., 2015) and ESPript 3.0 software were used (Robert and Gouet, 2014).
Detection of nuclear localization sequences (NLS) and hydrophobicity analysis
The search for putative NLS sequences, according to animal cell code, in the SARS-CoV-2 N and ORF6 proteins sequences was performed using WoLF PSORT tool (https://wolfpsort.hgc.jp/) (Horton et al., 2007), in order to identify possible putative regions targeting importins α (Fang et al., 2017).
Hydrophobic content analysis of ORF6 protein of SARS-CoV-2 and SARS-CoV was performed calculating GRAVY hydrophobicity index and constructing hydrophobicity plot according to Kyte and Doolittle index (Kyte and Doolittle, 1982). This analysis was performed in order to characterize the ORF6 protein of both viruses and to establish similarities in terms of possible transmembrane regions with α-helix structures (Fry et al., 2021).
Molecular docking protein-protein simulations
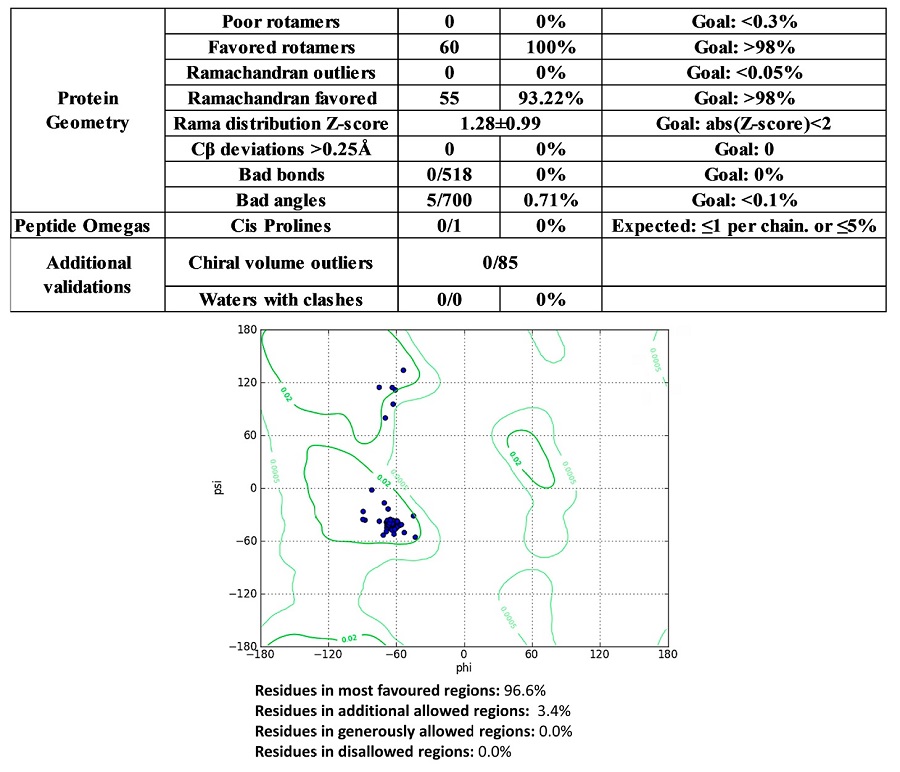
Molecular modeling of ORF6 protein structure (NCBI: QIV65092.1) was performed using Alphafold 2 (Jumper et al., 2021; Mirdita et al., 2022). Modeled structure was validated using MolProbity 4.5.1, PROCHECK and Chimera v. 1.16 software (Laskowski et al., 1993; Pettersen et al., 2004; Williams et al., 2018). The acceptance criteria for statistical and structural values are described in the Supplementary Figure S1.
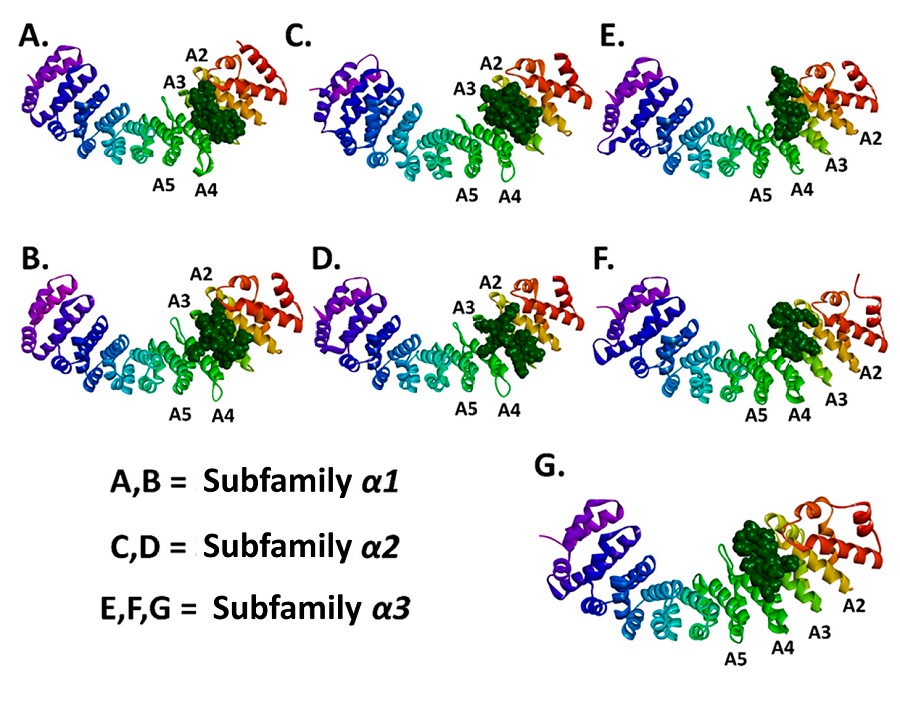
The molecular structure of SARS-CoV-2 N protein (PDB: 8FD5), and importins α isoforms of the α1 subfamily (isoforms α1 and α8), α2 subfamily (isoforms α3 and α4) and α3 subfamily (isoforms α5, α6 and α7) were obtained from Protein Data Bank RCSB database (https://www.rcsb.org/) and AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/) (Berman et al., 2000; David et al., 2022; Varadi et al., 2022) (Table 2).
Tabla 2. Estructuras de las isoformas de importina α utilizadas en esta investigación.
| Subfamily | Isoforms | Accession code | Database |
| α1 | Importin α1 | 4WV6 | Protein Data Bank RCSB |
| Importin α8 | A9QM74 | AlphaFold Protein Structure Database | |
| α2 | Importin α3 | 4UAE | Protein Data Bank RCSB |
| Importin α4 | O00505 | AlphaFold Protein Structure Database | |
| α3 | Importin α5 | P52294 | AlphaFold Protein Structure Database |
| Importin α6 | O15131 | AlphaFold Protein Structure Database | |
| Importin α7 | 4UAD | Protein Data Bank RCSB |
Molecular protein-protein docking simulations were performed employing Ibrahim et al. (2020) bioinformatic workflow, these assays were carried out between N protein and importin α isoforms, and between ORF6 protein and isoform importin α1, for it were used pyDock 3 software (Cheng et al., 2007). To select the complex with favorable binding affinities, were employed electrostatic, desolvation and van der Waals energies values (Cheng et al., 2007; Grosdidier et al., 2007; Jiménez-García et al., 2013; Pallara et al., 2017). Water molecules, ions and ligands were firstly removed from protein structures (Chook and Blobel, 2001).
Simulations were performed using receptor-ligand model generated by FTDock (Fourier Transform Dock) and pyDockRST algorithms (Chelliah et al., 2006; Cheng et al., 2007; Gabb et al., 1997; Grosdidier et al., 2007; Jiménez-García et al., 2013). Firstly, polar hydrogens were added at physiological pH 7.4, and also partial charges were incorporated to the protein structures using CHARMm force field, using the Discovery Studio Visualizer v. 20 software (Biovia Dassault Systemes, USA). Restraints distances methods were employed in importins α active sites (major binding site and minor binding site) corresponding to Armadillo (ARM) domains (ARM2, ARM3, ARM4, ARM6, ARM7, ARM8) (Chook and Blobel, 2001).
Resulting complexes were subjected to structural refinement process using 14.4 ps molecular dynamics simulation methods with GalaxyRefineComplex software (Heo et al., 2016). Free binding energy (ΔG) of complexes was calculated at 37° C (310.15 K) using PRODIGY software (Vangone et al., 2019).
Identification of active residues and intermolecular forces was performed using tridimensional (3D) and bidimensional (2D) representations, employing CHARMm force fields method with Discovery Studio Visualizer v. 20 software (Biovia Dassault Systemes, USA).
Molecular docking protein-ligand simulations
The molecular structure of ivermectin (ivermectin B1a) (PubChem CID Code: 6321424) was obtained from PubChem Database (https://pubchem.ncbi.nlm.nih.gov/) (Kim et al., 2016).
Energetic minimization of ivermectin structure was performed with Merck Molecular Force Field 94 (MMFF94) method, using for it, five steps per update with conjugate gradient algorithm, a total cycle number of 50000 steps, an energy convergence criteria of 0.001 kcal.mol−1.Å−1, also partial charges with Gasteiger force field and polar hydrogen atoms at physiological pH of 7.4 were added using Avogadro 2 software (Hanwell et al., 2012) and Chimera v. 1.16 software (Pettersen et al., 2004).
Molecular docking simulations between ivermectin and importin α isoforms were performed in a grid with dimensions of 43x73x78 Å3, 50 iterations in simulations and an exhaustiveness of 16, using PyRx software (Dallakyan and Olson, 2015) and Autodock Vina software (Trott and Olson, 2010).
Visualization of complexes, identification of binding site, active residues and intermolecular forces was performed employing CHARMm force fields and Momany-Rone partial charge, for it Discovery Studio Visualizer v. 20 software (Biovia Dassault Systemes, USA) was used.
Results and discussion
N proteins sequences analysis
Phylogenetic reconstruction reveals a close relationship between the SARS-CoV-2 N protein and SARS-CoV N protein sequences in a 94 % of the analysis (Figure 1A.).
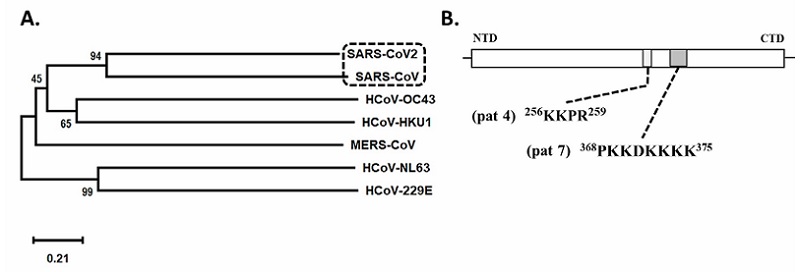
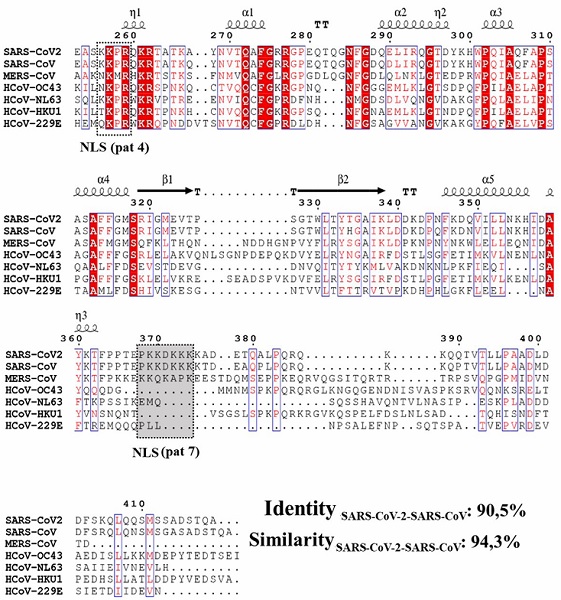
Results obtained in phylogenetic reconstruction are in agreement with genetic similarity reported in another research where an identity of 79.6 % to 80 % between both genomes was found (Ceraolo and Giorgi, 2020; Lu et al., 2020; Wu et al., 2020; Zhou et al., 2020). Kannan et al. (2020) and Ceraolo and Giorgi (2020) also described a high percentage of identity between amino acid sequences of nucleocapsid protein of SARS-CoV-2 and SARS-CoV, these authors suggest a 90 % to 90.52 % identity between these viral genomes, differences were registered in residues Gly25, Ser26, Asp103, Ala217 and Thr334, which were part of 380 residues substituted in the SARS-CoV-2 proteome (Wu et al., 2020) (Figure 2).
This sequences relationships suggests structural and functional similarities such as the ability to suppress Interferon Stimulation Response Element (ISRE) gene expression, that was also reported for other coronaviruses (Li et al., 2020; Kannan et al., 2020; Ceraolo and Giorgi, 2020; Chang et al., 2014; Timani et al., 2005) (Figure 1A.).
However, the analysis of SARS-CoV-2 N protein sequence showed the presence of a monopartite NLS sequence pat 4 “KKPR” (Lys-Lys-Pro-Arg) between residues 256-259, and a monopartite sequence of pat 7 “PKKDKKKK” (Pro-Lys-Lys-Asp-Lys-Lys-Lys-Lys) between residues 368-375 (Figure 1B.).
The NLS sequences detected in SARS-CoV-2 N protein are similar to those described in SARS-CoV N protein, however, in SARS-CoV a third NLS monopartite pat 7 sequence was recorded next to N-terminal domain between residues 38 to 44, which is absent in SARS-CoV-2 N protein due to substitution of residue 38 for a serine. The presence of NLS sequence next to the N-terminal domain of the SARS-CoV N-protein is functional, leading an exogenous protein into the nucleus, however it seems to be not relevant for the success of viral infection, becoming more important NLS sequences close to the C-terminal domain (Timani et al., 2005).
The similarity between SARS-CoV and SARS-CoV-2 N proteins is clearly visible in the multiple alignment representation, where a 90.5 % of identity and a 94.3 % of similarity between these two sequences were recorded (Figure 2).
Previous studies reported that SARS-CoV N proteins are cleaved during activation of caspases 3 and 7, that induce apoptosis in infected cells, generating fragments with functional NLS sequences that facilitate translocation of these fragments into the nucleus/nucleolus (He et al., 2004; Rowland et al., 2005; Timani et al., 2005).
It is important to mention that proteins fragments translocations into the nucleus occurs in low proportions at advanced stages of SARS-CoV infection, and that because the multimerization of N proteins blocks the cleavages (He et al., 2004; Timani et al., 2005). The presence of NLS sequences in SARS-CoV-2 N protein was also described by Gao et al. (2021).
Protein-protein molecular docking simulations of N protein and importin α isoforms
Molecular docking analysis evidenced binding affinities of the N proteins (NLS pat4, NLS pat7) to the ARM2-ARM4 domains of the evaluated importins α isoforms, these domains correspond to the major binding site, a NLS recognition site in importins (Baumhardt and Chook, 2018; Chook and Blobel, 2001) (Figure 3, Figure 4). It is important to denote that the entry of peptides/proteins with NLS sequences into the nuclear compartment occurs by classical importin α mediated pathway, NLS sequences possess the ability to interact with ARM domains of importins α (Chook and Blobel, 2001).
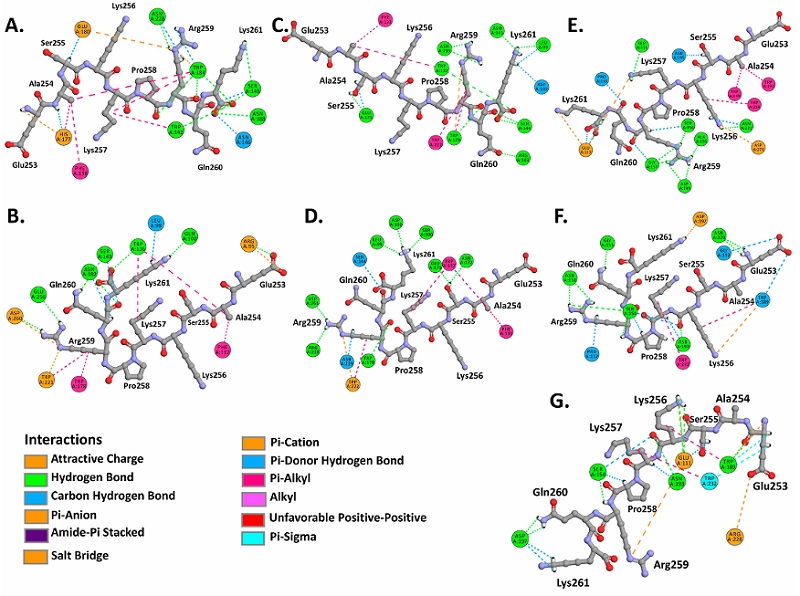
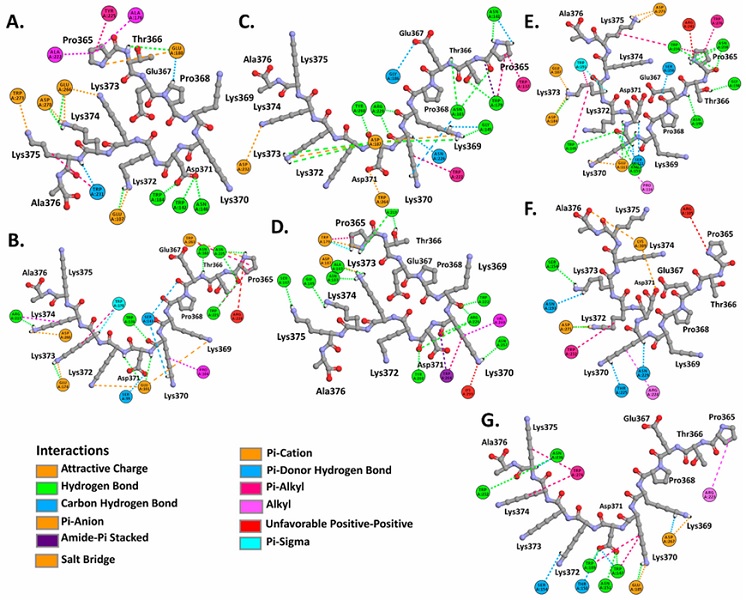
Resulting complexes between importins α isoforms and N protein (NLS pat4 and pat7) showed free binding energy values (∆G) ranging -10.0 to -6.3 kcal.mol-1, where the NLS pat7 sequence demonstrated the most favorable energy value, specifically to importin α3 and importin α5 (Figure 3B., Supplementary Table S1).
Active residues in complexes formed by N protein and isoforms of α1 subfamily were Phe138, Trp142, Asn146, Ser149, His177, Glu180, Trp184, Asn188, Asn228 in importin α1, and Arg95, Gln100, Glu107, Trp136, Ser143, Glu180, Asn182, Trp231, Glu256, Asp260, Glu266, Asp270, Trp273 in importin α8, residues that interact with NLS pat4 sequences through hydrogen bonds, unconventional interactions between a polarized carbon atom and hydrogen atom, interactions between Pi orbitals and donors groups of hydrogen bonds. The formation of electrostatic attractions and hydrophobic interactions were also identified (Figure 3A,B., Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
Residues Glu107, Trp142, Asn146, Ala176, Glu180, Trp184, Ala222, Tyr225, Trp231, Glu266, Asp270, Trp273 of importin α1, and residues Ser99, Glu101, Pro104, Trp136, Ser143, Arg217, Asp260, Trp263, Glu174, Trp178, Asn182, Trp221, Asn225 of importin α8, interact by hydrogen bonds, carbon hydrogen bonds, Pi interactions with hydrogen bonds donors, hydrophobic and electrostatic interactions with the NLS pat7 sequences (Figure 4A,B., Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
Complexes formed by α2 subfamily isoforms revealed as active residues to Leu99, Asp102, Arg103, Phe133, Trp137, Asn141, Ser144, Trp179, Glu175, Asn219, Trp222 in importin α3, Leu99, Ser100, Asp102, Phe133, Trp137, Ser144, Asn172, Glu175, Trp179, Arg218, Asn219, Trp222, Asp261 in importin α4, all of them interacting with the NLS pat4 sequences, exhibiting hydrogen bonds, carbon hydrogen bonds, electrostatic and hydrophobic interactions (Figure 3C,D., Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
Also registered as active residues were Trp179, Asn183, Gly186, Asp187, Asn219, Trp222, Asn226, Arg229, Asp232, Trp137, Asn141, Ala143, Gly145, Ser147, Asn257, Val260, Trp264, Tyr268 in importin α3, and Ala143, Gly145, Ser147, Trp179, Asn183, Asp187, Asn219, Trp222, Arg229, Asn257, Val260, Trp264, Tyr268 in importin α4, which interact with NLS pat7 sequences (Figure 4C,D., Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
Complexes registered with α3 subfamily isoforms showed as active residues to Ser111, Glu113, Pro116, Trp149, Ala155, Ser156, Gly157, Trp191, Asn195, Asp199, Asn231, Trp234, Asp273 of importin α5, and Pro112, Asn151, Ser154, Trp189, Gly192, Asn193, Asp197, Asn229, Trp232 of importin α6, and Glu111, Ser154, Trp189, Asn193, Asp197, Arg228, Trp232 in importin α7, all these residues interact with NLS pat4 sequences by hydrogen bonds, carbon hydrogen interactions, electrostatic attractions and hydrophobic interactions (Figure 3E,F,G,. Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
Also, residues Ser111, Glu113, Pro116, Trp149, Asn153, Ser156, Asp184, Glu187, Trp191, Asn195, Gly198, Trp234, Asn238, Asp273, Trp276 in importin α5, Ser154, Asn193, Thr225, Arg228, Asn229, Trp232, Asp271, Lys309 in importin α6, Trp147, Thr150, Asn151, Ser154, Glu185, Trp189, Arg223, Trp232, Asn236, Asp267, Trp274 in importin α7, were detected in interactions with NLS pat7 sequences (Figure 4E,F,G,. Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1).
ORF6 proteins sequences analysis
Amino acid sequences of SARS-CoV and SARS-CoV-2 ORF6 proteins showed 66.7 % of identity and 85.7 % of similarity among them (Figure 5A.).
Figure 5 A. Alignment of SARS-CoV and SARS-CoV-2 ORF6 protein sequences, importin α1 binding segment highlighted. B-C. Hydrophobicity plot and index of viral ORF6 according to Kyte & Doolittle (1982). B. SARS-CoV ORF6 protein. C. SARS-CoV-2 ORF6 protein.
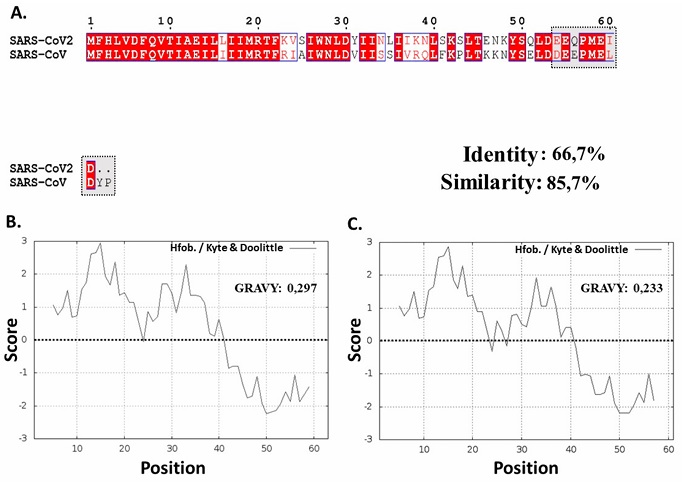
Figura 5. A. Alineación de las secuencias de las proteínas ORF6 de SARS-CoV y SARS-CoV-2, segmento de unión a importina α1 resaltado. B-C. Diagrama de hidrofobicidad e índice de ORF6 viral según Kyte & Doolittle (1982). B. Proteína ORF6 del SARS-CoV. C. Proteína ORF6 del SARS-CoV-2.
These proteins present high hydrophobicity scores in the Kyte and Doolittle scale (Kyte and Doolittle, 1982), evidencing hydrophobic regions in 72.13 % of the SARS-CoV-2 ORF6 protein, suggesting the presence of an α-helix transmembrane structure (between residues 1-44) (Hussain and Gallagher, 2010). The high content of residues with hydrophobic properties were evidenced in both proteins, which could also be verified with GRAVY hydrophobicity index that showed values of 0.297 and 0.233, respectively (Figure 5B,C.). The presence of SARS-CoV ORF6 protein in the membrane of the endoplasmic reticulum (ER)/ Golgi of the infected cell has been described in previous research; this similarity in terms of hydrophobicity indicators could suggest the same cellular localization of SARS-CoV-2 ORF6 and the same mechanism of action against importins α (Frieman et al., 2007). ORF6 proteins of SARS-CoV was also described with amphipathic characteristics, this protein mainly acts as antagonist of the host cell antiviral response and participates in the acceleration of infection kinetics (Frieman et al., 2007; Hussain and Gallagher, 2010; Liu et al., 2014; Zhao et al., 2009).
Due to their homology and high similarity, these proteins would be sharing functions mainly involved in the inhibition of ISRE promoter expression, so interferon beta (IFN-β) function is also affected (Kopecky-Bromberg et al., 2007), these observations were reconfirmed by Li et al. (2020) who reported an inhibition of interferon type I signaling pathway.
The complex importin α1-ORF6 SARS-CoV-2 protein showed a favorable free binding energy with a value of -7.7 kcal.mol-1. This complex demonstrated interaction of ORF6 protein C-terminal domain to the ARM2-ARM4 (major binding site) domains of importin α1 (Baumhardt and Chook, 2018) (Figure 6).
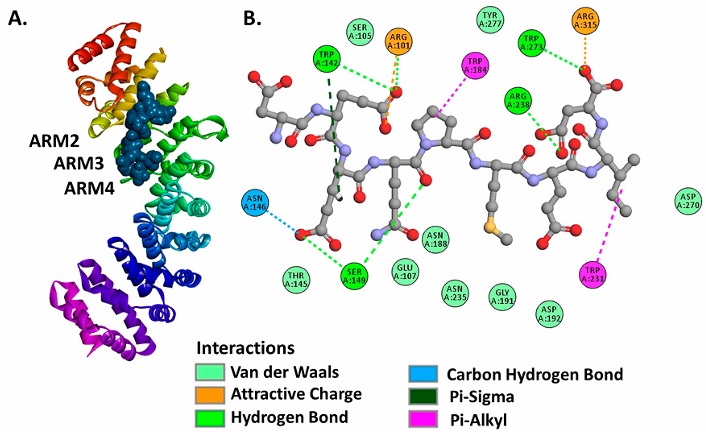
The active residues registered in importin α1 were Trp142, Arg101, Trp184, Arg238, Trp273, Arg315, Trp231, Ser149, Asn146 belonging to ARM2-ARM4 domains (Figure 6A.). The importin α1-ORF6 complex is stabilized with hydrogen bonds, carbon-hydrogen interactions, Pi orbitals and Sigma orbitals interactions, electrostatic attractions and hydrophobic interactions (Figure 6.B).
Studies revealed that SARS-CoV ORF6 protein is able to indirectly block STAT1/STAT2 proteins nuclear translocation, this occurs by interaction of this viral protein with importin α1, hijacking importin β1 from cytoplasm to ER/Golgi membrane surface (Frieman et al., 2007; Kopecky-Bromberg et al., 2007; Liu et al., 2014; Narayanan et al., 2008; Zhao et al., 2009). As described by Frieman et al. (2007) and Liu et al. (2014), the C-terminal domain (residues 54-63) of SARS-CoV ORF6 protein involved in the interaction to importin α1, is located within a region with similarity to residues 54-61 in SARS-CoV-2 ORF6 protein (Figure 5A.).
A recent study performed by Miyamoto et al. (2022), has revealed that SARS-CoV-2 ORF6 protein exhibits antagonistic effects on interferon mediated antiviral response by blocking intracellular traffic of STAT1 through interactions between viral protein and STAT1, and a high binding affinity to importins α1 of infected cells.
Another research revealed that SARS-CoV-2 ORF6 protein plays an important role in nucleocytoplasmic traffic, through an interaction to Rae1:Nup98 complex of the host cell, this event directly manipulate localization and functions of nucleoporins causing a damaged nucleocytoplasmic traffic, promoting the accumulation of RNA transporters (Addetia et al., 2021; Gordon et al., 2020; Kato et al., 2021).
Protein-Ligand molecular docking simulations of ivermectin and importin α isoforms
Molecular docking analysis of ivermectin and importin α isoforms, revealed favorable values of free binding energies ranging from -7.71±0.24 to -8.63±0.32 kcal.mol-1, showing a higher affinity mainly to ARM2-ARM4 domains that are topologically similar to those determined in the complexes formed by SARS-CoV-2 N protein and ORF6 proteins (Figure 3, Figure 4, Figure 7, Supplementary Table S1, Supplementary Table S2).
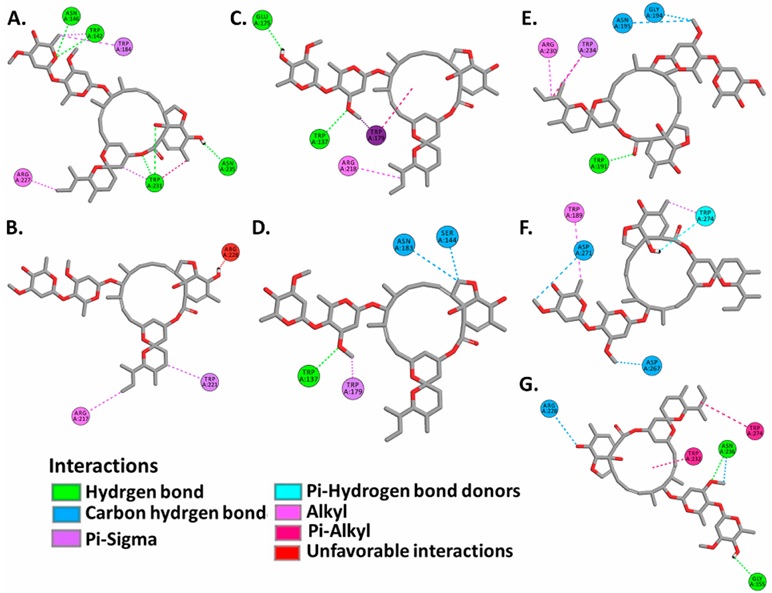
Active residues registered in α1 subfamily isoforms were Trp142, Asn146, Trp184, Arg227, Trp231, Asn235 in importin α1, Arg217 and Trp221 in importin α8, which interact with the ivermectin molecule through hydrogen bonds and Pi-Sigma interactions (Figure 7A,B,. Supplementary Table S2).
In complexes formed by α2 subfamily isoforms, active residues identified were Trp137, Glu175, Trp179, Arg218 in importin α3, and Trp137, Ser144, Asn183, Trp179 in importin α4, which interact with ivermectin by hydrogen bonds, carbon hydrogen bonds, Pi and Sigma orbitals interactions, as well as interactions between alkyl chains (Figure 7C,D,. Supplementary Table S2).
On the other hand, complexes formed with α3 subfamily isoforms revealed Trp191, Gly194, Asn195, Arg230, Trp234 as active residues in importin α5, Trp189, Asp271, Trp274 in importin α6, and Gly155, Arg228, Trp232, Asn236, Trp274 in importin α7. These residues interact with ivermectin by hydrogen bonds, interactions between polarized carbon atoms and hydrogen, alkyl chains interactions, Pi and Sigma orbitals interactions, Pi orbitals and alkyl chains interactions, and interactions between Pi orbitals and donors groups of hydrogen bond (Figure 7E,F,G,. Supplementary Table S2).
Human genome encodes seven importin α isoforms that are grouped into three subfamilies known as α1, α2 and α3, which share a common structure consisting in Importin β1 Binding domain (IBB) and ARM domains, that have the ability to interact with NLS sequences at major and minor binding sites, respectively (Pumroy and Cingolani, 2015).
Many of these isoforms have been extensively investigated due to their abilities to interact with viral proteins, this is the case of importin α1, which is a target to several viral proteins such as E1A of Adenovirus, N protein of Hantavirus, integrase and Vpr of HIV-1 (Fontes et al., 2000; Pumroy and Cingolani, 2015). In the same way, importin α3 is capable to interact with viral proteins such as PB2 polymerase of influenza A virus, capsid proteins of Chikungunya virus, E1 and E2 proteins of HPV virus (Bian and Wilson, 2010; Pumroy and Cingolani, 2015; Pumroy et al., 2015).
Other isoforms such as importin α5 and importin α7 form complexes with influenza A protein PB2; recently it was described that importin α6 and importin α7 interact with VP24 protein of Ebola virus (Tarendeau et al., 2007; Xu et al., 2014). On the other hand, α3 subfamily isoforms play an important role in STAT1/STAT2 heterodimers transport during the antiviral host response (Fagerlund et al., 2002; Sekimoto et al., 1997).
In the last decades it was reported that ivermectin is able to block nucleoplasm-cytoplasm traffic of viral proteins of DENαV, HIV-1, WNV, Hepatitis E Virus (HEV), Bovine Herpesvirus I (BoHV-1) and ZIKV avoiding their replicative cycle (Crump, 2017; Mastrangelo et al., 2012; Ōmura and Crump, 2014; Wagstaff et al., 2012; Yang et al., 2020). King et al. (2020) reported that ivermectin is also able to disrupt the interaction of human adenovirus viral E1A protein with importin α without disturbing importin α/β1 interactions.
An in vitro study performed by Caly et al. (2020), has evidenced that after 48-72 hours of treatment, ivermectin reduced about 5000 fold the presence of replicated SARS-CoV-2 genome in infected Vero/hSLAM cells, finding IC50 values between 2.2-2.8 µM, indicating a high antiviral activity.
Researchers suggest that this decrease could be occurring due to an inhibition of importin α/β1 complex activity, and was also hypothesized that ivermectin reacts between each other creating compound complexes that may interfere with viral processes (Caly et al., 2020; Rizzo, 2020).
Active residues identified in ivermectin-importin α complexes were also described in complexes mentioned above, so they could probably be sharing the same binding site (major binding site) in the evaluated isoforms (Chook and Blobel, 2001) (Figure 3, Figure 4, Figure 7, Supplementary Table S1, Supplementary Table S2). These observations are in agreement with Yang et al. (2020), who demonstrated in vitro that ivermectin has the ability to interact with ARM domains of importins α of Mus musculus, and may even cause instability of importin α/β1 complexes.
Although interactions between ivermectin and importin α are widely accepted, several computational studies have shown that this molecule could have other SARS-CoV-2 proteins as target. A recent in silico study performed by Azam et al. (2020), described that ivermectin demonstrated binding affinities to SARS-CoV-2 Nsp9 protein and to importin α, evidencing favorable interaction energies, however, importin α protein model used belongs to M. musculus.
In another study of biophysical and computational approach, was were reported binding affinities and interaction stability of ivermectin (avermectin B1a, avermectin B1b) with targets such as Importin α1 and Importin β1, as well as viral proteins such as helicase and protease MPRO, among which according to kinetic parameters evaluated show greater affinities to viral proteins (González-Paz et al., 2021).
Another computational research revealed that ivermectin exhibit higher binding affinities with the complex formed by RNA polymerase (RdRp) and SARS-CoV-2 RNA molecules, establishing ternary complex (Sen Gupta et al., 2022).
Another in silico study performed by Bello (2022), revealed that ivermectin could show as possible interaction target SARS-CoV-2 protease 3CLpro and Nsp9 protein. This was also registered by Choudhury et al. (2021), who reported favorable binding to SARS-CoV-2 protease, SARS-CoV-2 replicase and host cell TMPRSS2 (Transmembrane Serine Protease 2) protein. In agreement to this, another computational study performed by Saha and Raihan (2021) revealed that ivermectin may also be a suitable inhibitor of interactions between SARS-CoV-2 spike protein and human ACE2 (Angiotensin-Converting Enzyme 2) protein.
The findings presented in this study add to the current knowledge about the possible mechanisms of action of ivermectin against SARS-CoV-2 infection, which may result in the competition for the major binding site (ARM2-ARM4 domain) of the importin α isoforms by the viral proteins (N and ORF6) and the ivermectin molecules, allowing the decrease of viral replication success and the reduction of the inhibition of the antiviral response of host cells.
Conclusions
This research reports for the first time the identification of binding sites (ARM2-ARM4 domains) in human importin α isoforms shared by the N and ORF6 proteins of SARS-CoV-2 and ivermectin, suggesting it as one of the possible mechanisms of action of ivermectin against SARS-CoV-2 infection. This study leads to new fields of research focused on the exploration of new compounds with antiviral actions against COVID-19, that targets to this classical nucleocytoplasmic transport pathway involved in viral protein traffic and inhibition of antiviral response in infected cells.