











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
The activated phosphoinositide 3-kinase δ syndrome (APDS) [OMIM 615513] is an inborn error of immunity with an autosomal dominant inheritance pattern caused by a heterozygous pathogenic variant of the PIK3CD gene on chromosome 1p36, with a gain of function on its encoded product1,2. Its prevalence is estimated to be < 1: 1,000,000 live births3. The PIK3CD gene encodes the p110 delta isoform of the catalytic subunit of the activated phosphoinositide 3-kinase (PI3K) and is expressed in hematopoietic cells, participating in T and B lymphocyte homeostasis processes4-6.
APDS was first described in 20137. It has been reported to have incomplete penetrance and variable expressivity among patients, ranging from asymptomatic cases to cases with severe immunodeficiency8.
The diagnosis of APDS is suspected in patients with recurrent sinopulmonary infections, mainly bacterial and viral pneumonia, upper respiratory tract infections, rhinosinusitis, otitis, herpesvirus-associated infections, gastroenteritis, ocular infections, benign lymphoproliferation, and is confirmed by the detection of a pathogenic variant in the PIK3CD gene sequence9. The proposed treatment aims to prevent infections, limit lymphoproliferation, and control autoimmunity10.
The objective of this case report is to report the second case of APDS in Mexico. The information, figures, and biological samples presented were collected with the parents' and patient's previous written informed consent.
Clinical case
The case is a 17-year-old female with a family history of consanguinity between parents, systemic lupus erythematosus on both sides, and a healthy brother and sister.
At 9 months of age, she required hospitalization for pneumonia and presented with recurrent respiratory tract infections from the age of 4. From the age of 8, she had prolonged cough and purulent rhinorrhea and was diagnosed with bronchiectasis by computed tomography (CT) of the chest at 11 years old, which persisted in the most recent CT (Fig. 1).
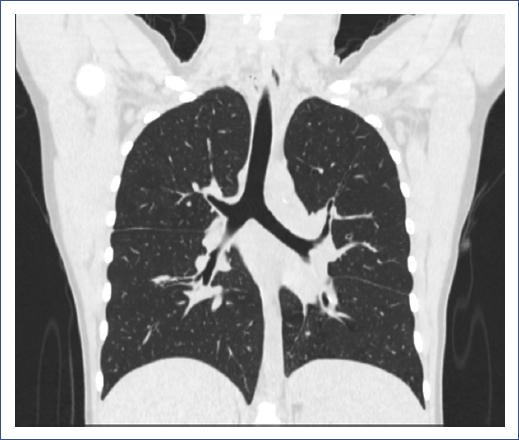
Figure 1 Computerized axial tomography at 17 years old, showing right upper solitary nodule, bilateral cylindrical lamellar atelectasis and bronchiectasis, left septal thickening, and axillary nodal hypertrophy.
At 12 years old, she presented with perforated otitis media requiring ventilation tube placement. At 16 years old, she underwent tonsillectomy and adenoidectomy due to the presence of unilateral tonsillar lymphoid hyperplasia, also presenting with pansinusitis, recurrent oral candidiasis, and chronic rhinitis (Fig. 2).
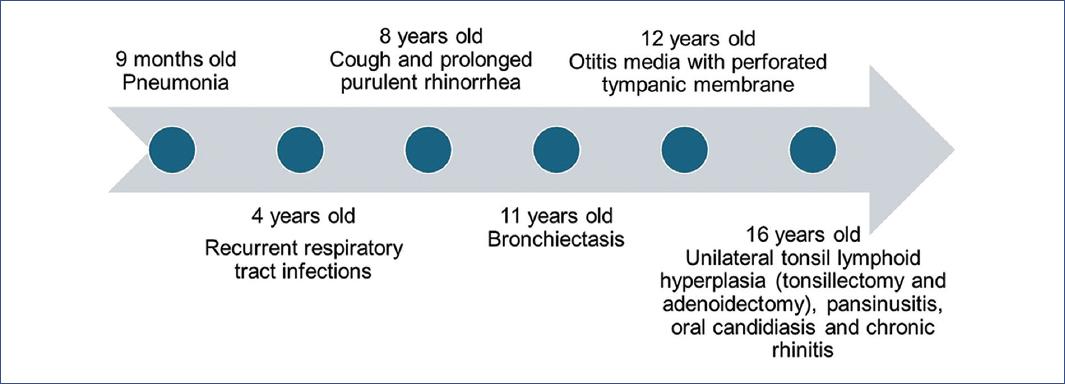
Figure 2 Clinical evolution with the most relevant findings that required medical attention throughout the patient's growth.
Physical examination revealed a weight of 56.9 kg (50th percentile), height of 1.65 m (65th percentile), non-obstructive septal deviation, turbinates with partial edema predominantly on the right, hypertrophy of lingual papillae, small, non-painful cervical lymphadenopathy, chest with generalized bilateral hypoventilated areas, and clubbing in upper and lower extremities.
Cystic fibrosis, immotile cilia syndrome, and autoimmunity were investigated and ruled out. In the presence of leukopenia, lymphopenia, low CD4 lymphocyte subpopulation, and immunoglobulin studies with persistently elevated immunoglobulin M (IgM) and normal levels of other immunoglobulins (Table 1), a probable Hyper IgM syndrome was suspected, and the patient was referred to the genetics department.
Table 1 Laboratory results
| Item | 24 October 2018 | 25 April 2019 | 17 June 2019 | 09 October 2022 |
|---|---|---|---|---|
| Leucocytes | ||||
| Patient value | 5770 cel/µL | 4497 cel/µL | 4480 cel/µL | |
| RR | 5000-10000 cel/µL | |||
| Total lymphocytes | ||||
| Patient value | 1080 cel/µL | 732.7 cel/µL | 776 cel/µL | 770 cel/µL |
| RR | 1000-3400 cel/µL | |||
| Immunoglobulin M | ||||
| Patient value | 297 mg/dL | 362 mg/dL | 3.140 mg/dL | 692 mg/dL |
| RR | 29-200 mg/dL | 0.40-2.30 mg/dl | 40-280 mg/dl | |
| C3 | ||||
| Patient value | 112 | 123 | - | 36.7 |
| RR | 90-180 | |||
| C4 | ||||
| Patient value | 16.3 | 17.9 | - | 6.39 |
| RR | 16-389 | |||
| Total CD3 cells | ||||
| Patient value | - | - | 526 cel/µL | - |
| RR | 600-3100 cel/µL | |||
| Total CD4 cells | ||||
| Patient value | 365 cel/µL | 385 cel/µL | 226 cel/µL | - |
| RR | 404-1612 cel/µL | 134-3061 cel/µL | 428-2020 cel/µL | |
| Total CD8 cells | ||||
| Patient value | 554 | - | 267 | - |
| RR | 259-1080 | 259-1080 | ||
| CD4/CD8 | ||||
| Patient value | 0.66 | - | 0.85 | - |
| RR | 1-2.9 | > 1 |
The bold highlighted values are outside of reference range. RR: reference range; cel/µL=cells/µL.
Next-generation sequencing and multiplex ligation-dependent probe amplification studies targeting inborn errors of immunity were performed, which reported a heterozygous pathogenic variant in the PIK3CD gene c.3061G>A (p.Glu1021Lys), confirming an autosomal dominant inborn error of immunity known as APDS. Treatment was initiated by the Immunology department with monthly intravenous immunoglobulin at a dose of 500 mg/kg of body weight, plus prophylactic antibiotics, with favorable evolution and better control of infectious processes. The pathogenic variant was screened for in the siblings using the same study but was not identified; the parents did not wish to know if either of them carried the variant.
Discussion
Our patient presented with pneumonia, recurrent respiratory tract infections, bronchiectasis, chronic rhinosinusitis, recurrent otitis media, tonsillitis requiring tonsillectomy, leukopenia, lymphopenia with low CD4 count, and persistently elevated IgM levels, which are clinical findings compatible with APDS10, confirmed by the identification of a heterozygous pathogenic variant in the PIK3CD gene c.3061G>A (p.Glu1021Lys). In this variant, there is a change from guanine to adenine at nucleotide 3061 of the coding DNA, which at the protein level generates a change from glutamic acid to lysine at amino acid 1021. This change causes a gain of function of the p110 delta catalytic subunit of the PI3K enzyme and corresponds to the most commonly reported variant in the literature11. The gain of function of the PIK3CD gene allows the coexistence of a state of immunodeficiency and autoimmunity12,13.
The PI3K protein family is formed by a heterodimeric protein with subunits p110-alpha (encoded by the PIK3CA gene), p110-beta (encoded by the PIK3CB gene), and p-110-delta (encoded by the PIK3CD gene), the latter associated with APDS114,15. In APDS, the gain of function of PIK3CD produces hyperactivity of p-110-delta, which hyperphosphorylates downstream mediators of mammalian target of rapamycin (mTOR), resulting in hyperactivation of the mTOR pathway. This alters B cell maturation and differentiation and increases the senescence of effector CD8 T cells, which are important for antiviral and antitumor functions. It also decreases the population of CD4 T cells and natural killer cells13,16,17, as well as causes dysgammaglobulinemia with increased serum IgM and deficiency of IgG and IgA17. Furthermore, it stimulates the proliferation of lymphocytes in lymph nodes, leading to lymphadenopathy7,18.
Due to the alterations in immune homeostasis described above, patients have increased susceptibility to infections by encapsulated bacteria, which affect up to 96% of patients with otitis media or rhinosinusitis7,9,19. About 80% of cases of pneumonia are bacterial, mainly caused by Haemophilus influenzae and Streptococcus pneumoniae; bronchiectasis occurs in 60% of cases; about 50% of patients present with chronic viral infections, mainly by Epstein-Barr virus and cytomegalovirus; fungal and parasitic infections are less frequent20. Up to 30% of patients with APDS develop autoimmune cytopenias21.
The treatment in these patients should be individualized, ranging from surveillance in asymptomatic cases to the use of prophylactic antimicrobials, immunoglobulin replacement, and even mTOR pathway inhibitors or haploidentical hematopoietic stem cell transplantation, which has been described to produce a reversal of the post-transplant phenotype22,23. At present, the use of selective PI3K-delta inhibitors is being studied, which is reported to have greater efficacy and fewer adverse effects, mainly in patients with lymphoproliferation23,24.
This case is the second reported in Mexico, as only one previous case was reported with the pathogenic variant PIK3CD c.3061G>A (p.Glu1021Lys) with de novo gain of function. This suggests that this change arises as a response to the presence of HIV in the parents to inactivate viral genes, and therefore, a probable relationship between infectious processes, immune response, and genetic changes should be studied25.
Diagnosing inborn errors of immunity, such as activated PI3K-delta syndrome, represents a challenge for current medicine due to the lack of recognition of the clinical data presented by patients and the low availability of molecular diagnostic studies. The patient's diagnosis in this case was prolonged, as it was not suspected during the years she remained under study. In particular, diagnosing activated PI3K-delta syndrome requires demonstrating the pathogenic variant of the PIK3CD gene through molecular studies that require per se clinical suspicion. Thus, publishing this case report provides a guideline to broaden the clinical suspicion that allows the integration of a targeted diagnosis of inborn errors of immunity.
Conclusion
This is the second case reported in the literature of APDS in Mexico, so it is important to know about it in order to make a timely diagnosis, for which a high clinical suspicion is required, in addition to immunological and genetic studies, in order to provide appropriate treatment and prevent complications.

