










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.83 no.1 Ciudad de México ene./mar. 2013
https://doi.org/10.1016/j.acmx.2013.01.010
Artículo de revisión
Las células T reguladoras en la enfermedad pulmonar obstructiva crónica
Regulatory T cells in chronic obstructive pulmonary disease
Leonardo Limón-Camachoª, Helena Solleiro-Villavicencioa, b, Ilana Pupko-Sissab, Ricardo Lascurainc y María Inés Vargas-Rojasb *
ª Unidad de Posgrado, Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), México, D.F., México.
b Departamento de Investigación en Tabaquismo y EPOC, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas (INER), México, D.F., México.
Correspondencia:
Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas,
Calzada de Tlalpan 4502, 14080 México, D.F., México.
Teléfono: +(52 55) 54871700 ext. 5305.
Correo electrónico: manecbba@yahoo.com (M.I. Vargas-Rojas).
c Departamento de Bioquímica, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas (INER), México, D.F., México.
Recibido el 1 de junio de 2012.
Aceptado el 16 de enero de 2013.
Resumen
La exposición al humo del tabaco induce inflamación de las vías aéreas y es el principal factor de riesgo para desarrollar la enfermedad pulmonar obstructiva crónica (EPOC). En este proceso inflamatorio participan varias poblaciones celulares. Algunas fallas en la modulación de la respuesta inflamatoria han sido aceptadas como un factor para el desarrollo de esta enfermedad. Las células T reguladoras (Treg) son un tipo de linfocitos T CD4+ que modulan la respuesta inmune mediante contacto directo con las células efectoras, así como por la secreción de citocinas inmunorreguladoras. El papel de las células Treg en la EPOC no se encuentra completamente comprendido, por lo cual es importante evaluar su participación en la inmunopatogénesis de la enfermedad.
Con el objetivo de elaborar una revisión sistemática de artículos originales que nos permitiera describir las células Treg (su origen, características y mecanismos de acción) y su participación en la EPOC, realizamos una búsqueda intencionada en las siguientes bases electrónicas: MEDLINE, AMED, PubMed y Scielo; para ello usamos la combinación de las siguientes palabras clave: <<COPD and Regulatory T cells/EPOC y células T reguladoras>>, <<Inflammation and COPD/Inflamación y EPOC>>, ''T regulatory cells/Células T reguladoras>>. Incluimos artículos de ciencia básica, ensayos clínicos controlados y no controlados, metaanálisis y guías.
A partir de esta búsqueda, concluimos que las células T reguladoras son una subpoblación de linfocitos T CD4+ cuyas funciones primordiales son la supresión de la respuesta inmune y el mantenimiento de la tolerancia a autoantígenos. Fallas en los mecanismos de regulación de las células T reguladoras conducen al desarrollo y perpetuación de la inflamación en la EPOC.
Palabras clave: Enfermedad pulmonar obstructiva crónica; Células T reguladoras; Células Th17; Inflamación; Inmunorregulación; México.
Abstract
Exposition to tobacco smoke has been established as the main risk factor to develop chronic obstructive pulmonary disease (COPD), by inducing inflammation of the airways. Several cell populations participate in this inflammatory process. It has been accepted that a maladaptive modulation of inflammatory responses plays a critical role in the development of the disease. Regulatory T cells (Treg) are a subset of T CD4+ lymphocytes that modulate the immune response through secretion of cytokines. The role of the Treg cells in chronic obstructive pulmonary disease is not clearly known, that is why it is important to focus in understanding their participation in the pathogenesis of the disease.
To elaborate a systematic review of original articles in which we could describe Treg cells (their ontogeny, mechanisms of action) and their role in COPD, we made a systematic literature search in some data bases (MEDLINE, AMED, PubMed and Scielo) looking through the next keywords: ''COPD and Regulatory T cells/EPOC y células T reguladoras'', <<Inflammation and COPD/Inflamación y EPOC>>, <<Regulatory T cells/Células T reguladoras>>. We included basic science articles, controlled and non-controlled clinical trials, meta-analysis and guides. From this search we conclude that Treg cells are a subpopulation of T CD4+ lymphocytes and their major functions are the suppression of immune responses and the maintenance of tolerance to self-antigens.
A disruption in the regulatory mechanisms of the Treg cells leads to the development and perpetuation of inflammation in COPD.
Keywords: Chronic obstructive pulmonary disease; T regulatory cells; Th17 cells; Inflammation; Immune regulation; Mexico.
Introducción
La exposición al humo del tabaco induce inflamación de las vías aéreas; en este proceso inflamatorio participa la respuesta de varias poblaciones celulares, como los neutrófilos, los macrófagos, los linfocitos y las células epiteliales1. Las interacciones entre estas poblaciones celulares son de particular importancia en el desarrollo del enfisema pulmonar. Aunque la asociación entre los mecanismos inflamatorios y la disminución en la función respiratoria no se conocen con claridad, algunos autores han sugerido una relación entre la gravedad del enfisema y la cantidad de células inflamatorias en los alvéolos2. Se ha observado una correlación entre el enfisema y el infiltrado celular en el tejido pulmonar, particularmente de los linfocitos T. Basándoses en la respuesta efectora y en los defectos de los procesos de activación y producción de citocinas observados en la enfermedad pulmonar obstructiva crónica (EPOC), se ha sugerido que hay un componente autoinmune que desencadena y provoca la cronicidad de la enfermedad3, o bien, que existen fallas en los mecanismos de regulación, ya sea por una falta de capacidad en las células reguladoras, o por una resistencia a la supresión en las células efectoras que causan la perpetuación de una respuesta inflamatoria4.
Uno de los mecanismos de regulación más estudiados es la supresión mediada por las células T CD4+CD25+FOXP3+, una subpoblación de células naturalmente anérgicas y supresoras que se denomina células T reguladoras (Treg).
El papel de las células Treg en la EPOC es controversial; Smyth et al. informaron que el fumar de forma crónica aumenta el número de células Treg en las vías aéreas5. Opuesto a estos resultados, 2 estudios informaron una disminución en los niveles de células Treg en pacientes con EPOC secundaria al tabaquismo, al compararse con los controles sanos6,7.
Es muy importante evaluar las células Treg en la EPOC, ya que estas podrían participar en la patogénesis de la enfermedad. Para comprender mejor el papel que ejercen las células Treg en la EPOC, en esta revisión se hará una descripción de las características de estas células y, posteriormente, se hablará sobre sus posibles mecanismos de acción en la enfermedad.
Métodos
Para comprender las características de las células Treg (origen, marcadores y mecanismos de acción) y el papel que ejercen en la EPOC se efectuó una revisión sistémica de los artículos originales más relevantes. Realizamos una búsqueda intencionada en las diferentes bases electrónicas: MEDLINE (101 resultados), AMED (144 resultados), PubMed (36.114 resultados) y Scielo (10.278 resultados), usando la combinación de las siguientes palabras clave: <<EPOC y células T reguladoras/COPD and Regulatory T cells>> (908 resultados), <<Inflamación y EPOC/Inflammation and COPD>> (7.277 resultados), <<Células T reguladoras/T regulatory cells>> (41.979 resultados).
Incluimos en esta búsqueda artículos originales de ciencia básica, ensayos clínicos controlados y no controlados, metaanálisis y guías. Se consideraron para esta investigación las publicaciones de trabajos en humanos y modelos animales, tanto in vivo como in vitro.
Los datos de esta revisión fueron extraídos y sintetizados dentro del reporte narrativo.
Desarrollo histórico del concepto de células T reguladoras
Después del descubrimiento en 1960 de las funciones de los linfocitos T cooperadores (Th, de T helper), Gershon y Kondo propusieron la existencia de una población de células con capacidad de suprimir la respuesta inmune8. Sin embargo, como en ese momento no se pudo identificar ningún marcador celular o alguna molécula soluble específica de dicha población, esta idea fue abandonada. En la década de los 70 se propuso que las células supresoras mediaban efectos biológicos al producir sustancias solubles. En la década de los 80, con la descripción de las células Th1 y Th2 se propuso que la supresión era el resultado de la actividad de las citocinas inmunorreguladoras producidas por estas subpoblaciones celulares9, por lo que fue descartada la existencia de una población reguladora. En esta misma década se demostró in vivo que los ratones timectomizados el tercer día de vida (d3Tx) desarrollaban enfermedades autoinmunes órgano-específicas, y que estas podían ser prevenidas con la administración de células T singénicas obtenidas del timo o del hígado de animales adultos10,11. En 1995 Sakaguchi et al.12 demostraron que la causa del desarrollo de estas enfermedades en los ratones d3Tx era la ausencia específica de una subpoblación de células T CD4+CD25+, y que su transferencia las prevenía; con esto se rescató el concepto propuesto por Gershon sobre la existencia de una población de células reguladoras.
Por otro lado, se descubrió que el Forkhead box P3, o factor de transcripción Scurfin (FOXP3), estaba altamente expresado en células T CD4+CD25+13. Previamente se había reportado en un modelo murino que la mutación del gen FOXP3 provocaba una deficiencia inmunológica causante de esplenomegalia, hepatomegalia, linfadenopatía y muerte temprana14, mientras que la mutación del gen ortólogo en el humano causa el síndrome de poliendocrinopatía inmune y enteropatía ligada al x (IPEX, las siglas en inglés de immunodysregulation, polyendocrinopathy, and enteropathy, x-linked syndrome)15,16. De esta manera se hizo evidente la participación del FOXP3 en el desarrollo y función de las células Treg.
Origen y desarrollo
El desarrollo de esta población celular se conoce como la tercera función del timo17, y requiere de la interacción del receptor de células T (TCR, siglas de T cell receptor) con las moléculas del complejo principal de histocompatibilidad de clase II (MHC II, siglas de major histocompatibility complex class II) unidas a un péptido propio expresado en el estroma tímico18. La expresión de FOXP3 es dependiente de la unión del CD28 con sus ligandos CD80/CD86 (fig. 1), y se requiere para que las células no sean eliminadas, a pesar del reconocimiento del antígeno con gran afinidad18. Lo anterior se ha demostrado en modelos donde hay una depleción de los genes FoxP3/FOXP3 (en los ratones y los humanos, respectivamente), y con la eliminación de las células Treg de los sujetos de estudio19. Por otro lado, se observó en modelos animales que aumentar la expresión de FoxP3 mediante la transducción de un retrovirus de este gen a células T CD4+CD25- les confiere la capacidad supresora20.
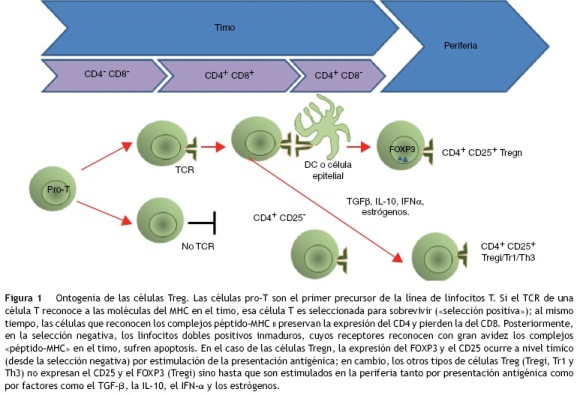
Caracterización fenotípica
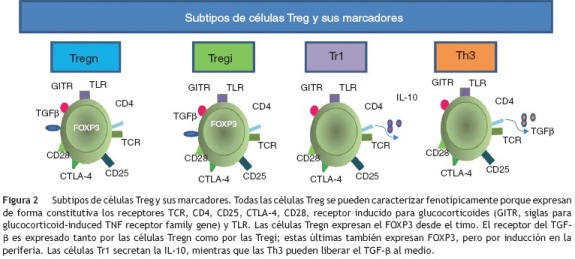
Se han descrito diferentes tipos de células reguladoras (tabla 1); las células Tr1 son un tipo de células Th dependientes de la interleucina (IL)-10 para su diferenciación, y algunas de sus propiedades reguladoras no expresan el FOXP3, pero al igual que las células T reguladoras naturales (Tregn), que son las que provienen del timo como células T CD4+CD25+FOXP3+, tienen alta expresión del CD152 en la superficie y pueden activar a la indolamina-2,3-dioxigenasa (IDO)21. Las células Th3 se caracterizan por la producción del factor de crecimiento transformante beta (TGF-β, siglas de transforming growth factor beta) y dependen de este para su diferenciación; además, expresan la cadena alfa del receptor de la IL-2 (CD25) como las células Tregn22. Otra población de células Treg son las inducidas (Tregi); estas tienen expresión de FOXP3 inducida en la periferia23.

Durante los últimos anos se ha buscado la manera de caracterizar a las células Treg; estas expresan de forma constitutiva el CD25, el CTLA-4 y el PD-L1, CD45RO, CD62L y CD12224,25; además, expresan receptores tipo Toll (TLR, siglas de Toll like receptors), entre los que están el 4, 5, 7 y 826. Todos estos marcadores son comunes a los expresados por células Th activadas, por lo que se han hecho numerosos experimentos con el intento de encontrar algún marcador exclusivo que las identifique. Las células Treg de los humanos expresan FOXP316 (fig. 2), que es considerado como el marcador fenotípico más importante, pues a pesar de que puede manifestarse en otras poblaciones celulares después de ser activadas, solo las células Treg tienen una expresión alta y sostenida del mismo.
La población de las células Treg aisladas del timo y la sangre periférica de los humanos representan entre el 5 y el 10% del total de los timocitos CD4+CD8- maduros, y del 1 al 2% del total de linfocitos T CD4+ en la sangre periférica24,25.
Mecanismos de acción
Se han propuesto diferentes mecanismos de acción por los cuales las células Treg median la supresión de las células efectoras (fig. 3); dentro de los principales están: a) un mecanismo dependiente de las citocinas, especialmente de la IL-10 y del TGF-p, en el que los factores solubles con características antiinflamatorias tienen una acción directa sobre la población de las células T efectoras, o bien un efecto inhibidor sobre las células presentadoras de antígeno, induciendo de esta manera una respuesta tolerogénica; b) un mecanismo de eliminación por medio de la vía de las granzimas-perforinas; c) un mecanismo de competencia, en el que las células Treg consumen los factores de crecimiento y supervivencia tales como la IL-2, que induce a la apoptosis de la población de las células efectoras, así como a la interferencia en sus mecanismos de activación; y, finalmente, d) un mecanismo dependiente del contacto celular, a través de la unión del CTLA-4 o el CD28 con sus ligandos CD80 y CD86.
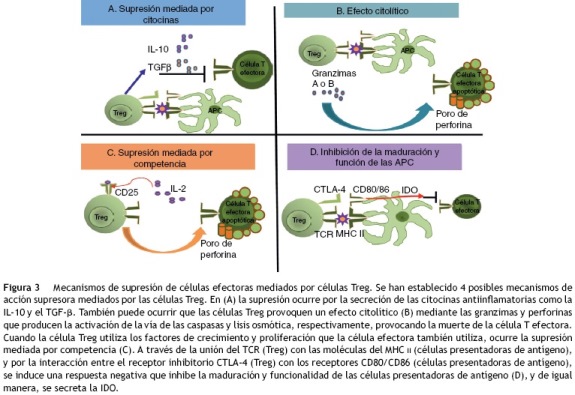
La participación de las citocinas, especialmente la IL-10, en la acción supresora de las células Treg fue ampliamente estudiada en la enfermedad inflamatoria intestinal de los modelos murinos; pero la participación de esta y del TGF-p es todavía controversial27. En los modelos in vitro, las células Treg humanas se activan a través de su TCR, y es necesaria una señal de coestimulación a través del CD2828. Se ha observado que al separar a las células Treg de las efectoras con membranas semipermeables, se evita la acción supresora, pues es a través del contacto celular que las células Treg se activan, y como estas tienen expresión constitutiva del CTLA-4, este compite con el CD28 por los sitios de unión; así, por la ausencia de una señal de coestimulación positiva, la presentación antigénica es insuficiente y se favorece un estado de anergia. Otro mecanismo molecular propuesto, también dependiente del contacto celular, es que la unión del CTLA-4 con su ligando inicia una vía de señalización negativa que resulta en que las células T que no proliferan, ya sea por efecto directo del contacto celular o bien por acción de factores humorales como la IL-10 o el TGF-p producidos por las células Treg. Algunos estudios demuestran que la interacción directa y el contacto entre células CD4+CD25- y las células Treg es necesario para la inmunorregulación. Por otro lado, se ha informado que las células Treg disminuyen la expresión del CD80 y el CD86 en las células presentadoras de antígeno29, lo que sugiere otro mecanismo de supresión. Adicional a la modulación directa o indirecta de la expresión del CD80 y el CD86, se producen señales que activan a la IDO en células dendríticas, una enzima que participa en el metabolismo del triptófano, en el cual se produce quinurenina, un mediador inmunosupresivo (fig. 3). Estas señales también promueven la localización de los factores de transcripción Foxo, que suprimen la expresión de genes que codifican para la IL-630.
Las células Tregn expresan a las granzimas A y B; además, tanto las células Tregn como las Tregi tienen citotoxicidad, dependiente de las perforinas, en contra de algunas células blanco (CD4+, CD8+, monocitos CD14+ y células dendríticas). Esta citotoxicidad es dependiente de las interacciones adhesivas producidas por el CD18, mas es independiente del complejo molecular Fas/FasL31. Cao et al.32 examinaron el papel de las perforinas y las granzimas en la eliminación de células tumorales. En su modelo experimental utilizaron líneas tumorales celulares que exclusivamente inducen la producción de la granzima B (pero no de la granzima A) en las células Treg; este experimento se realizó de esta manera, ya que las células Treg utilizan a la granzima B para la supresión de la eliminación tumoral mediada por las células NK y/o CD8+. La actividad supresora se ve reducida en las células Treg de ratones knock-out deficientes de la granzima B (Gzmb-/-); en consecuencia, los ratones Gzmb-/- eliminan los tumores de manera más eficiente que los ratones de tipo silvestre. Con este y otros experimentos se demostró que la actividad citolítica de las células Treg ejerce un papel importante en la supresión del mecanismo antitumoral mediado por las células NK y las T CD8+.
Las células Treg FOXP3+ pueden adquirir la capacidad de controlar específicamente la respuesta inmune de las células Th1, Th2 o Th17, ya que pueden modificar la expresión de los factores de transcripción específicos de cada linaje, T-bet, IRF-4 y ROR7t, respectivamente; estos factores de transcripción pueden influir en los patrones de expresión de los receptores de las quimiocinas, que facilitan la migración de las células Treg a un sitio particular de la inflamación33-35.
Es importante señalar que la actividad supresora de las células Treg debe estar controlada, ya que si fuera constante, podría afectar la capacidad del sistema inmune de combatir infecciones o el cáncer. Para explicar esto Pasare y Medzhitov describieron un modelo de bloqueo de las células Treg mediado por los TLR36; el bloqueo de la actividad supresora de las células Treg resulta de una activación de las células dendríticas, de tal manera que es posible una respuesta inmune adaptativa patógeno-específica. Un modelo similar propuesto por Sutmuller et al.37 demostró que las células Treg son inactivadas durante las infecciones, cuando los productos microbianos se unen al TLR2.
Partiendo de estos nuevos hallazgos, es posible hablar de las células Treg como una subpoblación más del tipo CD4+, y no como un linaje dedicado exclusivamente a la supresión. Se ha propuesto que poseen la capacidad de activar funciones del sistema inmune, por ejemplo la secreción de la IgA por parte de las células B38. Es decir, que las células Treg in vivo modulan la respuesta inmune de diferentes maneras, dependiendo, posiblemente, del estímulo, del lugar, del microambiente de citocinas y, al parecer, de su presencia en el momento en que las células T efectoras reciben el estímulo de activación39.
Características inflamatorias en la enfermedad pulmonar obstructiva crónica
La EPOC se caracteriza por la existencia de una respuesta inflamatoria pulmonar anormal hacia partículas nocivas o gases40; la exposición aguda a fuentes dañinas como el humo de cigarro provoca la activación de diversos receptores de reconocimiento de patrones (PRR, siglas de pattern-recognition receptor). Los PRR se activan de manera directa por las moléculas presentes en el humo de cigarro, o de manera indirecta por el daño celular que provocan dichas moléculas, produciéndose los patrones moleculares asociados a daño (DAMP, siglas de danger-associated molecule pattern). Se ha encontrado que en el lavado broncoalveolar (LBA) de pacientes con la EPOC existe un aumento en la concentración de DAMP como el ácido úrico, las proteínas de alta movilidad41, así como el ATP extracelular42. En un modelo experimental se demostró que la inhibición de los receptores purigénicos P2 (que se unen al ATP extracelular) previene el enfisema pulmonar inducido por el humo del cigarro43.
En las células pulmonares, otra respuesta al daño celular es la autofagia. En pacientes con la EPOC desde el estadio cero, se sabe que existe un nivel elevado de vacuolas autofágicas (autofagosomas y autolisosomas)44.
Las células que desempeñan un papel importante en la fase efectora de las respuestas inflamatorias de las inmunidades innata y adaptativa son los neutrófilos, los macrófagos, las células dendríticas y los linfocitos T, que secretan mediadores químicos como elastasas, meta-loproteinasas de matriz celular (MMP, siglas de matrix metalloproteinases) y citocinas. Algunos estudios han revelado que en el LBA y en el músculo liso de las vías aéreas de pacientes con la EPOC se encuentra aumentado el número de neutrófilos45. Estos estimulan la secreción del moco al favorecer la expresión de la mucina mediante la producción de la elastasa y las especies reactivas de oxígeno46.
Los macrófagos también secretan algunas MMP, particularmente la MMP9, así como otro tipo de proteasas, como las colagenasas 1 y 2, y las gelatinasas A y B47. Los niveles de macrófagos también se encuentran aumentados en el esputo y el LBA de los pacientes con la EPOC48. Se cree que los macrófagos son la fuente principal de las MMP en las vías aéreas; estas sustancias tienen la capacidad de degradar todos los componentes de la matiz extracelular, por lo que son los factores que más contribuyen a la destrucción del pulmón y al daño de las vías aéreas.
Se ha asociado con el consumo de cigarro una expansión de la población de las células de Langerhans en el epitelio del tracto respiratorio bajo. Posterior a la exposición aguda al humo de cigarro, las células dendríticas mieloides son reclutadas, y se han encontrado en el LBA de los sujetos fumadores49. Al comparar el LBA de los fumadores con el de los sujetos que nunca han fumado, se observa en el primero un incremento en la expresión de la langerina y el CD1a, ambos marcadores de células de Langerhans; asimismo, se ha cuantificado una mayor cantidad de moléculas de coestimulación (CD80 y CD86) en las células dendríticas mieloides procedentes del LBA de los fumadores. Este incremento también pudo ser observado en diversas poblaciones de células dendríticas del parénquima pulmonar de sujetos con la EPOC, y que el aumento en la expresión de las moléculas de coestimulación correlacionaba con la severidad de la enfermedad50,51.
Los estudios relacionados con los linfocitos T se han enfocado en la participación de los linfocitos T CD8+. Experimentos recientes han mostrado un incremento en el número de linfocitos T CD8+ en las biopsias bronquiales de pacientes con la EPOC, al compararse con las de fumadores sin EPOC52. Ensayos con determinados ratones knock-out han sugerido que estas células ejercen un papel crítico, ya que se ha observado que los ratones CD8-/- no desarrollan el enfisema tras ser expuestos al humo del cigarro53. Los mecanismos por los cuales estas células actúan para favorecer el proceso inflamatorio aún permanecen inciertos. Un posible mecanismo de acción es por citotoxicidad mediada por las granzimas y perforinas; esta vía se ha puesto de manifiesto en el esputo y en el LBA de pacientes con la EPOC, pues en estas muestras se ha encontrado un incremento en la cantidad de las perforinas54. Otros mecanismos de acción son la liberación de algunas citocinas proinflamatorias como la IL-13, la IL-17 y la IL-1855, y el reclutamiento de macrófagos56.
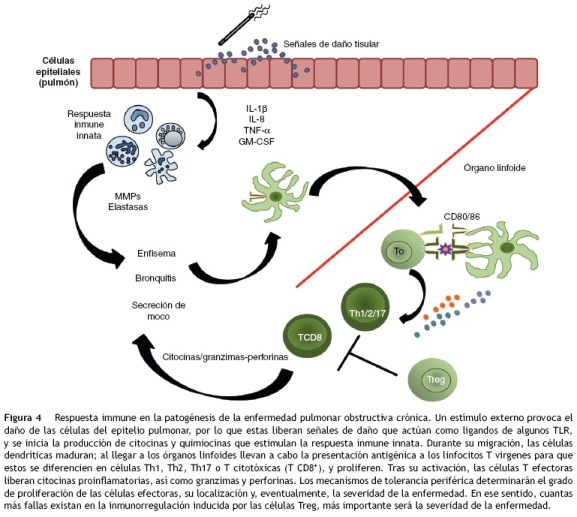
Se ha observado que el número de células T CD4+ aumenta en las vías aéreas y la sangre periférica de los pacientes con la EPOC4,57, y que el incremento es significativo a los 30 años de consumir tabaco58. Las células Th2 secretan la IL-4 y la IL-13, y es mediante esta última citocina que estimulan la producción del moco59. Por su lado, las células Th1 son productoras del IFN-7, que permite la activación de los macrófagos60. Se ha encontrado un incremento en el número de las células Th17 presentes en muestras de sangre periférica tanto de sujetos fumadores como de pacientes con la EPOC; asimismo, se sabe que existe una correlación negativa entre la cantidad de las células Th17 y el grado de obstrucción pulmonar (medido por el FEV1 y la relación FEV1 /FVC)4. Estas células han sido identificadas por el papel crítico que juegan en el desarrollo inflamatorio que ocurre en las enfermedades autoinmunes61. Su nombre deriva de la capacidad que poseen de producir la IL-17A y la IL-17F, que son citocinas que no son secretadas por las células Th1 y Th262,63. La importancia de estas células en el desarrollo de la EPOC es que la IL-17 estimula a las células epiteliales, los fibroblastos de las vías aéreas y las células del músculo liso para que secreten las quimiocinas que favorecen el reclutamiento de los neutrófilos. La falla en los mecanismos de regulación de los linfocitos Th17 conduce a la perpetuación de la respuesta inflamatoria en la EPOC4 (fig. 4).
Las células B también se encuentran aumentadas en los pacientes con la EPOC, según lo observado en las biopsias bronquiales. Se han encontrado cúmulos de células B en las vías aéreas pequenñas64, así como en el parénquima pulmonar de pacientes con la EPOC65, particularmente en los estadios 111 y iv de GOLD.
Tal como se ha observado en otros tejidos, los folículos linfoides de los pacientes con la EPOC son el resultado de una neogénesis linfoide66 y pertenecen al tejido linfoide asociado a bronquios, un tejido linfoide ectópico que se forma en los procesos inflamatorios67; este tejido recluta antígenos de las vías aéreas, inicia respuestas inmunes locales, y mantiene células de memoria en los pulmones; también es aquí en donde ocurre el cambio de isotipo de las inmunoglobulinas65. El papel de las células B en la EPOC ha sido controversial, pues por un lado su función puede ser benéfica en contra de las infecciones del tracto respiratorio; en contraste, se habla de un rol negativo cuando los anticuerpos van dirigidos contra componentes del tejido pulmonar, sugiriendo un componente autoinmune en la patogénesis de la EPOC, particularmente en el enfisema68.
Células T reguladoras en la enfermedad pulmonar obstructiva crónica
La presencia de las células Treg en los pacientes con la EPOC ha sido estudiada mediante citometría de flujo; los resultados obtenidos a partir de esta técnica han demostrado que existe un aumento de las células Tregn en la sangre periférica de pacientes con la EPOC en estado de exacerbación, y que el incremento está correlacionado con la gravedad de la enfermedad69. Asimismo, se ha observado que esta población celular se encuentra aumentada en el tejido pulmonar de los pacientes con enfisema secundario al consumo de tabaco70. De acuerdo con los resultados obtenidos a partir del LBA de los pacientes con la EPOC, se ha determinado que existe una correlación positiva entre el número de células CD4+CD25+ y el índice tabáquico (paquetes/ano)5, en tanto que en los conductos aéreos gruesos de los pacientes con la EPOC se ha observado un incremento en la expresión del FOXP3 que se correlaciona positivamente con el grado de consumo de tabaco (índice tabáquico)71. Al analizar los folículos linfoides, que están aumentados por la enfermedad, se observó un incremento en la expresión del FOXP3 en las células T CD4+ de pacientes con la EPOC. Sin embargo, las células Treg presentes en los folículos son de los tipos Tregi o Th3 más que del tipo Tregn, ya que la formación de células Tregi se ve favorecida por la presentación antigénica subóptima en un microambiente que contiene citocinas promotoras del desarrollo de estas poblaciones celulares; por ejemplo, las células epiteliales alveolares tipo II proveen un microambiente rico en TGF-p que favorece el desarrollo de células Th3. Las células Tregi se caracterizan por tener una expresión inestable del FOXP3, por este motivo se piensa que las células reguladoras presentes en los folículos linfoides de los pacientes con la EPOC son del tipo inducido, y que la inestabilidad en la expresión del FOXP3 podría estar reduciendo su capacidad reguladora72.
En ensayos realizados por Roos-Engstrand et al.73 se observó que la reducción en la expresión del FOXP3 en el LBA de los sujetos fumadores con función pulmonar normal indica que el aumento en la expresión del CD25 en las muestras no está asociado con la expansión de las células Treg. En cambio, la alta expresión del CD127 y la menor expresión del CD25 en los fumadores y en los pacientes con la EPOC moderada implican un predominio de las células T efectoras CD25+ que no poseen capacidad reguladora; en este estudio también se encontró que en sujetos ex fumadores (con más de 5 años sin fumar) es posible restablecer las proporciones normales de las subpoblaciones de los linfocitos T.
Algunos cambios epigenéticos podrían estar implicados en la función de las células reguladoras en la EPOC, por ejemplo, el funcionamiento de las histonas desacetilasas74. Por otro lado, la reducción en los niveles de la IL-10 en los pacientes con la EPOC al compararse con controles sanos72 sugiere otro mecanismo por el cual la inmunorregulación no es eficiente en este grupo de pacientes.
Conclusiones
Las células Treg son una subpoblación de linfocitos T CD4+ cuyas funciones primordiales y más estudiadas son la supresión de la respuesta inmune y el mantenimiento de la tolerancia a los autoantígenos. Estas células expresan de manera constitutiva marcadores que no son exclusivos de este linaje celular. Recientemente se ha incorporado FOXP3 como un marcador fenotípico característico de las células Treg, ya que en estas se expresa de manera elevada y sostenida, además de ser un factor crítico para el desarrollo y función de esta clase de linfocitos.
Se han descrito algunos mecanismos moleculares mediante los cuales actúan las células Treg. Concluimos que la modulación de la respuesta inmune que ejercen las células Treg ocurre a través de distintas vías, por lo que es importante evaluar bajo qué condiciones se activa una u otra.
La EPOC se caracteriza por una respuesta inflamatoria exagerada hacia partículas nocivas y gases. El sistema inmunorregulador en la EPOC no actúa de manera eficiente en la prevención del daño pulmonar y de las vías aéreas; por este motivo, hay un predominio de las respuestas proinflamatorias. Se ha demostrado que existe un aumento de las células Treg en la sangre periférica, el LBA y los cortes histológicos de pulmón de pacientes con EPOC, y que el incremento está correlacionado con la gravedad de la enfermedad. Fallas genéticas y epigenéticas podrían explicar la deficiente actividad inmunorreguladora de las células Treg en la EPOC; sin embargo, también es probable que las células efectoras presenten resistencia a ser reguladas; esto debe ser evaluado en trabajos futuros.
Financiación
No se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. Hodge S, Hodge G. Flow cytometric characterization of cell populations in bronchoalveolar lavage and bronchial brushings from patients with chronic obstructive pulmonary disease. Cytometry B Clin Cytom. 2004;61:27-34. [ Links ]
2. Domagala-Kulawik J. Effects of cigarette smoke on the lung and systemic immunity. J Physiol Pharmacol. 2008;59:19-34. [ Links ]
3. Núnez B, Sauleda J, Antó JM, et al., PAC-COPD Investigators. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183:1025-31. [ Links ]
4. Vargas-Rojas MI, Ramírez-Venegas A, Limón-Camacho L, et al. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Med. 2011;105:1648-54. [ Links ]
5. Smyth IJ, Starkey C, Vestbo J. CD4-regulatory cells in COPD patients. Chest. 2007;132:156-63. [ Links ]
6. Lee SH, Gosawami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567-9. [ Links ]
7. Barceló B, Pons J, Ferrer JM, et al. Phenotypic characterisation of T-lymphocytes in COPD: Abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J. 2008;31:555-62. [ Links ]
8. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: The role of thymic lymphocytes. Immunology. 1970;18:723-37. [ Links ]
9. Levings MK, Sangregorio R. Human CD25+CD4+ regulatory T cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295-302. [ Links ]
10. Mosmann TR, Coffman RL. Th1 and Th2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145-73. [ Links ]
11. Nishizuka Y, Sakukura T. Thymus and reproduction: Sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science. 1969;166:753-5. [ Links ]
12. Sakaguchi S, Fukuma K, Kuribayashi K, et al. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subsets a possible cause of autoimmune disease. J Exp Med. 1985;181:72 -87. [ Links ]
13. Viguier M, Lemaitre F, Verola O, et al. FoxP3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the junction of infiltrating T cells. J Immunol. 2004;173:1444-53. [ Links ]
14. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057-61. [ Links ]
15. Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20-1. [ Links ]
16. Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430-5. [ Links ]
17. Seddon B, Mason D. The third function of the thymus. Immunol Today. 2000;21:95-9. [ Links ]
18. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12:157-67. [ Links ]
19. Khattri R, Cox T. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337-42. [ Links ]
20. Loser K, Hansen W, Apelt J, et al. In Wtro-generated regulatory T cells induced by Foxp3-retrovirus infection control murine contact allergy and systemic autoimmunity. Gene Ther. 2005;12:1294-304. [ Links ]
21. Munn DH. Indoleamine 2,3-dioxygenase, Tregs and cancer. Curr Med Chem. 2011;18:2240-6. [ Links ]
22. Litjens NH, Boer K, Betjes MG. Identification of circulating human antigen-reactive CD4+ FOXP3+ natural regulatory T cells. J Immunol. 2012;188:1083-90. [ Links ]
23. Chen Y, Adams E, Regateiro FS, et al. Activation rather than Foxp3 expression determines that TGF-β-induced regulatory T cells out-compete naïve T cells in dendritic cell clustering. Eur J Immunol. 2012;42:1436-48. [ Links ]
24. Baecher-Allan C, Brown JA, Freeman GJ, et al. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245-53. [ Links ]
25. Dieckmann D, Plottner H, Berchtold S, et al. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J Exp Med. 2001;193:1303-10. [ Links ]
26. Wild CA, Brandau S, Lindemann M. Toll-like receptors in regulatory T cells of patients with head and neck cancer. Arch Otolaryngol Head Neck Surg. 2010;136:1253-9. [ Links ]
27. Stope MB, Rönnau C, Schubert T, et al. Transforming growth factor β in prostate cancer: Cellular effects and basic molecular mechanisms. Urologe A. 2012;51:1692-6. [ Links ]
28. Gogishvili T, Lühder F, Goebbels S, et al. Cell-intrinsic and -extrinsic control of Treg-cell homeostasis and function revealed by induced CD28 deletion. Eur J Immunol. 2012;43:188-93. [ Links ]
29. Zhou H, Li WM, Zhang M, et al. Foxp3-transfected CD4+ CD25- T cells suppress function of dendritic cells. Zhongguo Shi Yan Xue YeXue Za Zhi. 2008;16:164-9. [ Links ]
30. Kerdiles YM, Stone EL, Beisner DR, et al. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890-904. [ Links ]
31. Grossman WJ, Verbsky JW, Barchet W, et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589-601. [ Links ]
32. Cao X, Cai SF, Fehniger TA, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635 -46. [ Links ]
33. Koch MA, Tucker-Heard G, Perdue NR, et al. The transcription factor T-bet controls regulatory T cell homeostasis function during type 1 inflammation. Nat Immunol. 2009;10:595-602. [ Links ]
34. Zheng Y, Chaudhry A, Kas A, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351 -6. [ Links ]
35. Zhou L, Lopes JE, Chong MM, et al. TGF-beta-induced Foxp3 inhibits TH17 cell differentiation by antagonizing ROR-yt function. Nature. 2008;453:236-40. [ Links ]
36. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell mediated suppression by dendritic cells. Science. 2003;299:1033-6. [ Links ]
37. Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485-94. [ Links ]
38. Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740-50. [ Links ]
39. Vargas-Rojas MI, Crispin JC, Richaud-Patin Y, et al. Quantitative and qualitative normal regulatory T cells are not capable of inducing suppression in SLE patients due to T-cell resistance. Lupus. 2008;17:289-94. [ Links ]
40. Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2011. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease. Medical Communications Resources. [ Links ]
41. Ferhani N, Letuve S, Kozhich A, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:917-27. [ Links ]
42. Lommatzsch M, Cicko S, Muller T, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928-34. [ Links ]
43. Cicko S, Lucattelli M, Muller T, et al. Purigenic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185:688-97. [ Links ]
44. Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autop-hagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3:e3316. [ Links ]
45. Blidberg K, Palmberg L, Dahlen B, et al. Increased neutrophil migration in smokers with or without chronic obstructive pulmonary disease. Respirology. 2012;17:854-60. [ Links ]
46. Takeyama K, Dabbagh K, Jeong S, et al. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: Role of neutrophils. J Immunol. 2000;164:1546 -52. [ Links ]
47. Segura-Valdez L, Pardo A, Gaxiola M, et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684-94. [ Links ]
48. Dominguez-Fandos D, Peinado VI, Puig-Pey R, et al. Pulmonary inflammatory reaction and structural changes induced by cigarette smoke exposure in the Guinea pig. COPD. 2012;9:473 -84. [ Links ]
49. Lommatzsch M, Bratke K, Knappe T, et al. Acute effects of tobacco smoke on human airway dendritic cells in vivo. Eur Respir J. 2012;35:1130-6. [ Links ]
50. Freeman CM, Martinez FJ, Han MK, et al. Lung dendritic cell expression of maturation molecules increases with worsening chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180:1178-88. [ Links ]
51. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol. 2009;10:1237-44. [ Links ]
52. Roos-Engstrand E, Pourazar J, Behndig AF, et al. Cytotoxic Tcells expressing the co-stimulatory receptor NKG2 D are increased in cigarette smoking and COPD. Respir Res. 2010;11:128. [ Links ]
53. Maeno T, Houghton AM, Quintero PA, et al. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol. 2007;178:8090-6. [ Links ]
54. Hodge S, Hodge G, Nairn J, et al. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD. 2006;3:179-87. [ Links ]
55. Imaoka H, Hoshino T, Takei S, et al. Interleukin-18 production and pulmonary function in COPD. Eur Respir J. 2008;31:287-97. [ Links ]
56. Borchers MT, Wesselkamper SC, Harris NI, et al. CD8+ T cells contribute to macrophage accumulation and airspace enlargement following repeated irritant exposure. Exp Mol Pathol. 2007;83:301-10. [ Links ]
57. Chen C, Shen Y, Ni CJ, et al. Imbalance of circulating T-lymphocyte subpopulation in COPD and its relationship with CAT performance. J Clin Lab Anal. 2012;26:109-14. [ Links ]
58. Majo J, Ghezzo H. Lymphocyte population and apoptosis in the lungs of smokers and their relation with emphysema. Eur Respir J. 2001;17:946-53. [ Links ]
59. Whittaker L, Niu N, Temann UA, et al. Interleukin-13 mediates a fundamental pathway for epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol. 2002;27:593-602. [ Links ]
60. Kang MJ, Lee CG, Cho SJ, et al. IFN-gamma-dependent DNA injury and/or apoptosis are critical in cigarette smoke-induced murine emphysema. Proc Am Thorac Soc. 2006;3:517-8. [ Links ]
61. Pan HF, Leng RX, Feng CC, et al. Expression profiles of Th17 pathway related genes in human systemic lupus erythematosus. Mol Biol Rep. 2013;40:391-9. [ Links ]
62. Li HJ, Zhang CQ, Yu CX, et al. Roles of Th17 lymphocytes and inflammatory cytokines in airway inflammation exacerbation of murine asthmatic model. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2012;28:1126-38. [ Links ]
63. Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123-32. [ Links ]
64. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645-53. [ Links ]
65. Demoor T, Bracke KR, Maes T, et al. Role of lymphotoxin-alpha in cigarette smoke-induced inflammation and lymphoid neogenesis. Eur Respir J. 2009;34:405-16. [ Links ]
66. Brusselle GG, Demoor T, Bracke KR, et al. Lymphoid follicles in (very) severe COPD: Beneficial or harmful? Eur Respir J. 2009;34:219-30. [ Links ]
67. Randall TD. Bronchus-associated lymphoid tissue (BALT) structure and function. Adv Immunol. 2010;107:187-241. [ Links ]
68. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445-54. [ Links ]
69. Xiong XZ, Jin Y, Zhou Q, et al. Correlation between FoxP3+ regulatory T cells and chronic obstructive pulmonary disease. Zhonghua Yi Xue Za Zhi. 2008;88: 471-4. [ Links ]
70. Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567-9. [ Links ]
71. Isajevs S, Taivans I, Strazda G, et al. Decreased FOXP3 expression in small airways of smokers with COPD. Eur Respir J. 2009;33:61 -7. [ Links ]
72. Plumb J, Smyth JC. Role of regulatory T-cells in chronic obstructive pulmonary disease. Annals of Respiratory Medicine. 2009;33:61 -7. [ Links ]
73. Roos-Engstrand E, Pourazar J, Behndig AF, et al. Expansion of CD4+CD25+ helper T cells without regulatory function in smokingand COPD. Respir Res. 2011;12:74. [ Links ]
74. Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967-76. [ Links ]

