











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La infección del tracto urinario (ITU) es de las infecciones bacterianas más comunes a nivel mundial. Su clasificación difiere de acuerdo con el enfoque clínico. La guía de infecciones urológicas de la Asociación Europea de Urología (EAU, por sus siglas en inglés) las agrupa en cinco tipos: ITU no complicada, complicada, recurrente, asociada a catéteres y urosepsis (Tabla I), (Bonkat et al., 2021).
Tabla I Clasificación de ITU dentro de la Guía de Infecciones Urológicas adoptada por la Asociación Europea de Urología (Traducido y modificado de Bonkat et al., 2021).
| Clasificación de ITU | Descripción |
|---|---|
| ITU no complicada | ITU aguda esporádica o recurrente que se presenta en vías inferiores (cistitis no complicada) y/o altas (pielonefritis no complicada). Limitado a pacientes sin anomalías anatómicas y funcionales relevantes conocidas dentro del tracto urinario o comorbilidades. |
| ITU complicada | ITU en pacientes con mayor probabilidad de un curso complicado, es decir, infecciones asociadas a factores que comprometen el tracto urinario o la defensa del huésped, incluyendo en éstas la obstrucción urinaria, la retención urinaria causada por enfermedades neurológicas, inmunosupresión, falla renal, embarazo y la presencia de cuerpos extraños como los cálculos renales. |
| ITU recurrente | Término referido indistintamente a ITU complicada y/o no complicada, cuya frecuencia es de tres episodios por año o dos episodios en los últimos seis meses. |
| ITU asociada a catéter | ITU en personas cuyo tracto urinario se encuentra cateterizado o ha tenido un catéter colocado en las últimas 48 hrs. |
| Urosepsis | Disfunción multiorgánica que compromete la vida, causada por una respuesta desregulada del huésped ante una ITU. |
Por otro lado, a las ITU también se les clasifica por sus características epidemiológicas y el lugar donde se adquiere la infección que puede ser: la comunidad (CAUTIs), el ambiente intrahospitalario (HAUTIs) y asociadas al ambiente hospitalario con inicio de síntomas en la comunidad (CO‑HAUTIs). Las más comunes son las CAUTIs, afectan a más de 150 millones de personas al año (Öztürk & Murt, 2020). Sin embargo, la cantidad está subestimada debido a que la ITU no es de notificación obligatoria en los centros de atención de salud primaria a nivel mundial.
En el año 2018, la Encuesta Nacional De Atención Médica Ambulatoria (NAMCS) reportó un total de 3,577 millones de casos de ITU en los EE. UU. Además, diversos autores estiman que cada año más de 7 millones de personas suelen acudir a las instituciones de atención médica por ITU, lo que representa el 0.7% de los servicios médicos ambulatorios (Tandogdu & Wagenlehner, 2016; Taur & Smith, 2007).
En el año 2022, Yang, Chen, Zheng, Qu, Wang & Yi, realizaron una recopilación y análisis de datos globales de las ITU y el resultado fue un incremento del 60.4% en el número de casos a nivel mundial en el año 2019 con respecto al año 1990. Ellos, reportan un aumento del 140.2% en las muertes globales asociadas con las ITU, ya que en el año 1990 fueron un total de 98,590 con respecto a las 236,790 en el año 2019. En su estudio mencionan un crecimiento de su presencia en pacientes de la tercera edad y el problema de que del 40 al 75% de los tratamientos con antibióticos son inadecuados.
En lo que respecta a Latinoamérica, existe una carencia de datos sobre las ITU. No obstante, en México, el Sistema Nacional De Vigilancia Epidemiológica (SINAVE) informa de un total de 3,231,627 casos en el año 2022 ubicándolas en el segundo lugar de las veinte principales causas de enfermedad nacional (Figura 1), (SINAVE, 2024).
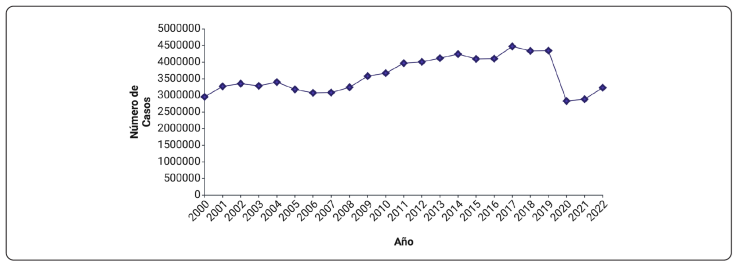
Creación propia realizada en BioRender.com
Figura 1 Reporte de casos anuales de las infecciones del tracto urinario (Reporte Anual 2000-2022. SINAVE).
Es común que a las ITU se les relacione con la aparición de cepas resistentes y a la incapacidad de erradicar completamente a los microorganismos, por esto es importante el conocimiento sobre la variación genética de los diferentes agentes etiológicos para llevar a cabo un tratamiento antimicrobiano adecuado (Abou-Heidar, Degheili, Yacoubian & Khauli, 2019). Al mismo tiempo, el conocimiento sobre la resistencia a los antibióticos en Latinoamérica, en particular en México, se limita a estudios de pequeños grupos de investigación u hospitales con un restringido número de muestras (Ballesteros-Monrreal et al., 2023; Barrios-Villa, Picón, Reynaga & Arenas-Hernández, 2023; de Zavaleta et al., 2015).
Una ITU puede ser por microorganismos del tubo digestivo del paciente, donde abundan los bacilos gram negativos como Escherichia coli. A pesar de ser parte de la microbiota intestinal, algunas cepas de E. coli son capaces de causar una enfermedad extraintestinal como la ITU, por lo que recibe el nombre de E. coli uropatógena (UPEC) que es reconocida como el principal agente etiológico de esta importante enfermedad (Koga et al., 2014; Torres & Mattera, 2006).
Diversos estudios en Colombia, Argentina y México han encontrado a UPEC como el agente etiológico predominante en la ITU con una incidencia de más del 70% (Ambuila-González, Ramírez-López, Escobar-Bedoya & Chávez, 2015; Ballesteros-Monrreal et al., 2023; Córdova et al., 2014; Orrego, Henao & Cardona, 2014).
La vigilancia epidemiológica de las cepas UPEC en México, se basa principalmente en la determinación de los perfiles de virulencia y de resistencia al evaluar la presencia y/o ausencia de biomarcadores genéticos mediante pruebas fenotípicas, pruebas de susceptibilidad antimicrobiana y moleculares, como la reacción en cadena de la polimerasa (Polymerase Chain Reaction o PCR). Se han utilizado otros métodos como la electroforesis en gel de campos pulsados (Pulsed Field Gel Electrophoresis o PFGE) para la determinación de huellas dactilares del ADN Multi-locus y su tipificación de secuencias (Multi-locus Sequence Typing o MLST) para establecer una relación clonal entre los aislados. Sin embargo, el uso de la secuenciación del genoma completo (Whole Genome Sequencing o WGS) ha proporcionado un análisis más profundo y detallado de la filogenia, virulencia y resistencia de las cepas de UPEC, lo que permite la identificación y caracterización de cepas emergentes, su origen y la identificación de cepas portadoras de genes de resistencia y virulencia, y la ubicación de estos en el genoma bacteriano.
La WGS ha revolucionado los enfoques microbiológicos tradicionales para ofrecer un método más confiable para caracterizar las cepas clínicas de UPEC. Por ello, el objetivo de esta revisión es conocer el impacto del análisis de la WGS como un complemento de otros métodos de estudio para entender el papel de UPEC como patógeno.
Escherichia coli es un bacilo Gram negativo, anaerobio facultativo, mide aproximadamente 1.1-1.5 µm por 2.0-6.0 µm. pertenece a la familia Enterobacteriaceae. E. coli se puede diferenciar serológicamente mediante tres antígenos principales de la superficie celular: el O (somático), H (flagelar) y K (capsular).
La mayor parte de las cepas de E. coli están en la microbiota intestinal. No obstante, algunas han evolucionado a través de la adquisición de diversos genes que codifican para factores de virulencia, que les confieren una mayor adaptación a nuevos nichos y provoca una amplia gama de enfermedades (Kaper, Nataro & Mobley, 2004).
Las cepas de E. coli que tienen la capacidad de generar una enfermedad se dividen en dos grupos principales: E. coli diarreogénica (DEC) y E. coli extraintestinal (ExPEC), (Figura 2).
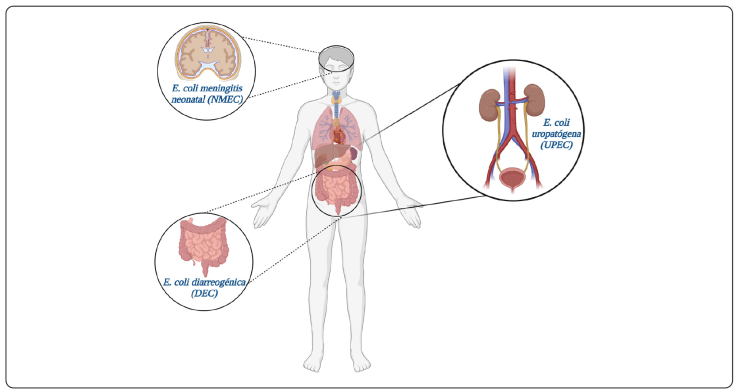
Las cepas de ExPEC, provocan diversas enfermedades y son las ITU las más frecuentes con una alta incidencia a nivel mundial donde UPEC es el agente etiológico principal responsable de más del 80% de los casos.
Las cepas de UPEC generalmente tienen una cantidad heterogénea de factores de virulencia que lo convierten en uno de los patógenos más exitosos en el desarrollo de una ITU (Barrios-Villa et al., 2023). UPEC tiene la capacidad de utilizar diversos entornos nutricionales como el intestino, la vejiga, los riñones y el torrente sanguíneo, ya que su metabolismo se encuentra altamente regulado y es sensible a la disponibilidad de nutrientes, que le permiten sobrevivir en entornos competitivos y fluctuantes de nutrientes (Mann, Mediati, Duggin, Harry & Bottomley, 2017).
Diversidad genética de E. coli
E. coli es uno de los microorganismos más utilizados en el laboratorio por su versatilidad y facilidad de manejo, es el modelo microbiológico más importante dentro de la biología molecular, ya que ha sido una pieza medular para el desarrollo de la fisiología y genética bacteriana (Blount, 2015). Por la versátil y diversidad de su genomas es multifacético capaz de colonizar tanto a humanos como animales; esto se aprecia en las diferentes combinaciones de factores de virulencia que varían de acuerdo con el nicho ecológico y/o hospedero en el que se encuentre. Por ello, los diferentes patotipos de E. coli se encuentran asociados a: condiciones clínicas, la epidemiología, la filogenia y los factores de virulencia diferentes (García & Fox, 2021; Leimbach, Hacker & Dobrindt, 2013).
La versatilidad genómica de E. coli ha sido revelada gracias a la WGS. Antes de la introducción de esta herramienta, su estructura clonal se determinaba mediante la serotipificación de cepas clínicas, cuya variabilidad serológica no era aleatoria, ya que algunos serotipos están asociados a patotipos específicos (Ingle et al., 2016; Ørskov & Ørskov, 1992). La electroforesis enzimática multi -locus (Multilocus Enzyme Electrophoresis o MLEE) apoyó la teoría de que la variabilidad de E. coli no se debe a una selección natural, sino a su aptitud bacteriana (Tenaillon, Skurnik, Picard, & Denamur, 2010). La WGS también reveló su plasticidad genómica, producto de su capacidad para cambiar, adquirir o perder genes, y de la selección natural, lo que resulta en su diversidad genética y fenotípica (Blount, 2015).
Serotipificación
La serotipificación es una herramienta clave en el análisis bacteriano y, desde una perspectiva epidemiológica, facilita la identificación de la prevalencia de una serovariedad en diferentes regiones geográficas. En el caso de E. coli, la serotipificación se basa en el uso de anticuerpos de conejo1 generados contra los diferentes tipos de antígenos O y H. E. coli presenta una gran diversidad en su antígeno O, lo que lo convierte en un biomarcador importante para su clasificación serológica. Esta diversidad ha hecho de la serotipificación el estándar de oro para la tipificación de E. coli. La combinación de los antígenos O y H (por ejemplo, UPEC O25:H4) son fundamentales para la detección de brotes epidemiológicos, la diferenciación taxonómica de E. coli, la identificación de serotipos patógenos dentro de la especie, así como para estudios clonales y evolutivos (García & Fox, 2021).
Se han descrito al menos 186 antígenos O, 56 antígenos H y 80 antígenos K. El uso de los antígenos O y H es suficiente para identificar a la mayoría de las cepas de E. coli. Emplear el antígeno O contribuye no sólo a determinar el serogrupo, y la combinación de este con el antígeno H, sino también el serotipo (Fratamico, DebRoy, Liu, Needleman, Baranzoni & Feng, 2016; Tenaillon et al., 2010).
El serotipo es considerado un marcador de tipos o linajes específicos de E. coli, ya que el número de posibles combinaciones de antígenos O y H conocidos, de esta bacteria, es superior a 10 000. Se han reportado un gran número de eventos de recombinación alrededor del locus del antígeno O, la región está sujeta a una fuerte presión de selección de los sistemas inmunitarios, de los hospederos mamíferos y a la depredación de los bacteriófagos. Por tanto, es posible que dos aislados estrechamente relacionados tengan serotipos diferentes, o que dos aislados de E. coli de linajes no relacionados hayan convergido en el mismo serotipo (Ingle et al., 2016).
E. coli uropatógena se ha asociado a un número limitado de serogrupos (O1, O2, O4, O6, O7, O8, O16, O18, O25, O62 y O75) y serotipos (O1:H4, O1:H6, O1:H7, O1:H-, O2:H1, O2:H4, O4:H5, O6:H1, O7:H4, O7:H6, O7:H-, O18ac:H7, O18ac:H-, O22:H1, O25:H1, O75:H5 y O75:H7), (Arenas-Hernández, Navarro-Ocaña, Molina-Villa, Martínez-Alvarado, Aroche-Camarillo & Martínez-Laguna, 2011). Sin embargo, se han encontrado nuevos serotipos asociados a las cepas de E. coli que producen infecciones urinarias, aunque es importante mencionar que un gran porcentaje de estas son no tipificables (NT), (Ballesteros-Monrreal et al., 2020).
La serotipificación tradicional tiene limitaciones como su elevado costo, la presencia de reactividad cruzada de los antisueros, algunos serogrupos, y la variación de anticuerpos entre lotes. Es por ello que se desarrolló una metodología molecular para la determinación del serogrupo y el serotipo. A diferencia de la serotipificación tradicional que utiliza antisueros producidos contra los diferentes tipos O y H de E. coli, la serotipificación molecular generalmente se refiere a ensayos basados en la genética que se dirigen a genes específicos dentro del operón que codifica al antígeno O y a genes del antígeno H que codifican los diferentes tipos de antígenos flagelares (Fratamico et al., 2016; Ingle et al., 2016).
Por su estructura variable, se han reportado hasta 181 antígenos O y algunos de ellos se dividen en subgrupos, como el O9 (O9 y O9a), O18 (O18ab y O18ac), O28 (O28ab y O28ac) y O112 (O112ab y O112ac), (Liu et al., 2020).
La determinación genotípica del serotipo se basa en genes relacionados a la biosíntesis de los antígenos O y H. Para el antígeno O se usan los genes wzx (codifica para una flipasa), wzy (codifica para una polimerasa), wzm y wzt (codifican para los transportadores ABC). La determinación del antígeno H se basa principalmente en los genes fliC, fllA y flmA que (codifican para flagelina); también se usa el gen flkA que (codifica para una proteína gancho), (Bessonov et al., 2021; Iino et al., 1988; Ratiner, 1998).
La WGS ha permitido una mayor precisión en la identificación del serotipo mediante pruebas in silico. El Serotype Finder, es una herramienta web del Centro de Epidemiología Genómica (Epidemiology Genomic Center o CGE que para efecto de lo anterior utiliza el genoma completo y la base de datos de los genes del antígeno O (wzx, wzy, wzm y wzt) y del antígeno H (fliC, flkA, fllA, flmA y flnA) que está en constante actualización (Joensen, Tetzschner, Iguchi, Aarestrup & Scheutz, 2015). Por consiguiente, la WGS da mejores resultados con respecto a la tipificación tradicional, ya que se puede determinar si existen mutaciones puntuales en los genes utilizados para su identificación o si hay discrepancias. Asimismo, se ha observado que los antígenos O y H tienen alelos que son muy similares entre sí, un ejemplo de esto son los antígenos O17, O44, O73, O77 y O106 los cuales comparten una similitud del 99% en los alelos de los genes wzx y wzy. Los antígenos H4 y H17 coinciden en un 97-99% de similitud en el gen fliC (Bessonov et al., 2021).
Tipificación de secuencias multi-locus (MLST)
La tipificación de secuencias multi-locus (MLST, por sus siglas en inglés) utiliza secuencias completas o fragmentos internos de genes de mantenimiento2 (usualmente seis o siete genes/ fragmentos) que están sujetos a escasa presión selectiva. Estas secuencias/fragmentos son catalogados de acuerdo con su variación alélica y se les asigna un tipo de secuencia o linaje de acuerdo con la combinación alélica observada en cada uno de los genes (Maiden et al., 1998; Urwin & Maiden, 2003).
Para llevar a cabo la MLST de una cepa bacteriana en el laboratorio se amplifican los siete u ocho genes mediante PCR, se someten a secuenciación y se realiza la comparación de los perfiles alélicos para determinar su secuencia tipo (ST), (Figura 3) (Uelze et al., 2020). Los genes utilizados en los esquemas MLST son: adk (adenilato kinasa), fumC (fumarato hidratasa), gyrB (ADN girasa), icd (Isocitrato deshidrogenasa), mdh (Malato deshidrogenasa), purA (Adenilsuccinato sintetasa), y recA (Motivo de unión ATP/GTP) para el esquema de Achtman y, dinB (ADN polimerasa IV), icdA (Isocitrato deshidrogenasa), pabB (p-aminobenzoato sintasa), polB (ADN polimerasa II), putP (Prolin permeasa), trpA (subunidad α de la triptófano sintasa), trpB (subunidad β de la triptófano sintasa), uidA (β-glucuronidasa) para el esquema Pasteur. Este método es capaz de proporcionar datos para la vigilancia epidemiológica y su análisis requiere la interpretación detallada de los datos de secuenciación obtenidos de los genes de mantenimiento y la información generada a partir de una serie de herramientas informáticas como las “variaciones alélicas” proporcionadas por el Centro de Epidemiología Genómica (https://cge.food.dtu.dk/services/MLST/) y la del Instituto Pasteur (https://bigsdb.web.pasteur.fr/ecoli/), (Jaureguy et al., 2008).
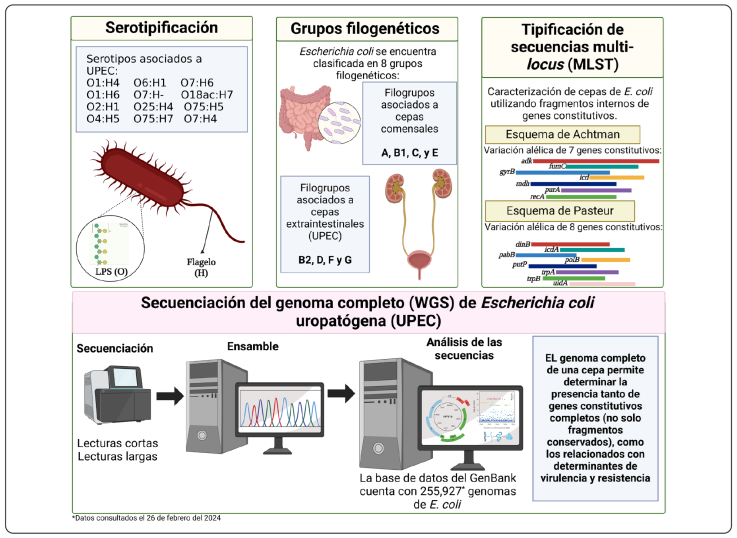
Creación propia realizada en BioRender.com
Figura 3 Análisis integral para el estudio de E. coli. UPEC. Escherichia coli uropatógena; LPS. Lipopolisacárido; O. Antígeno somático; H. Antígeno flagelar; adk: adenilato kinasa; fumC: fumarato hidratasa; gryB: ADN girasa; icd: isocitrato deshidrogenasa; mdh: malato deshidrogenasa; purA: adenilosuccinato sintetasa; recA: motivo de unión ATP/GTP; dinB: ADN polimerasa; icdA: isocitrato deshidrogenasa; pabB: p -amniobenzoato sintasa; polB: polimerasa II; putP: prolin permeasa; trpA: subunidad A de la triptófano sintasa; trpB: subunidad B de la triptófano sintasa; uidA: Beta-glucuronidasa (Clermont et al., 2013; Genome-NCBI-NLM, n.d.; Kauffmann, 1947).
La herramienta del Centro de Epidemiología Genómica, denominada MLST 2.0, con una base de datos amplia almacena la información en cinco bases de datos online. Esta información es recolectada por la Universidad de Oxford mediante una base de datos pública llamada PubMLST útil para la tipificación molecular y el estudio de la diversidad del genoma bacteriano (Jolley, Bray & Maiden, 2018; Larsen et al., 2012). La plataforma MLST 2.0 cuenta con alrededor de 70 esquemas MLST para 66 especies bacterianas diferentes. Esta herramienta se encuentra en actualización constante mediante un script3 automatizado para la descarga mensual de todas las secuencias de alelos y perfiles de ST de las bases de datos del PubMLST. Para la determinación de la ST, se sube en la plataforma el genoma de interés, se específica la bacteria que se está trabajando y la plataforma realiza un BLAST para la búsqueda en el genoma de todos los alelos de los genes que conforman el MLST. En el caso de E. coli se cuentan con dos esquemas MLST: el esquema 1, el cual utiliza 7 genes (esquema de Achtman) y el esquema 2 que emplea 8 genes (esquema de Pasteur). Una vez identificados los alelos, se determina la ST que se basa en la combinación resultante de estos alelos (Larsen et al., 2012). El uso de herramientas web de libre acceso que se encuentran en constante actualización permite un resultado más específico en la asignación de la ST.
Filogenia
E. coli es un microorganismo diverso presente en la mayoría de los mamíferos del mundo. Algunas cepas de E. coli, como se mencionó, tienen la capacidad de causar enfermedades intra y extraintestinales tanto en humanos como en animales (Clermont, Bonacorsi, & Bingen, 2000; Ørskov & Ørskov, 1992). Al estudiar la diversidad de las cepas patógenas de esta bacteria mediante técnicas de electroforesis enzimática multi-locus 4 (MLEE, por sus siglas en inglés) y ribotipificación5, encontraron la existencia de subestructuras genéticas asociadas con su sitio de aislamiento. Los primeros ensayos filogenéticos determinaron la presencia de 4 grupos filogenéticos principales (A, B1, B2 y D) y dos accesorios (C y E), (Selander, Caugant, Ochman, Musser,Gilmour, & Whittam, 1986; Herzer, Inouye, Inouye, & Whittam, 1990; Tenaillon et al., 2010). Posteriormente, con el desarrollo de herramientas moleculares como PCR, Clermont et al. (2000) desarrollaron una PCR triple para poder llevar a cabo la clasificación filogenética de las cepas de una manera más rápida y eficaz. La PCR triple consiste en la determinación de 3 genes, el gen chuA (involucrado en el transporte del grupo hemo), yjaA (presente en E. coli K-12, función desconocida) y TspE4.C2 (fragmento de ADN) (Clermont et al., 2000).
En 2008, Jaureguy et al., al trabajar con 161 cepas de E. coli obtenidas de bacteriemias, e identificar los polimorfismos de los nucleótidos obtenidos de la secuenciación de 8 genes de mantenimiento (dinB, icdA, pabB, polB, putP, trpA, trpB y uidA), encuentran algunas inconsistencias entre la PCR triple propuesta por Clermont et al (2000) y los filogrupos obtenidos en su trabajo. Al hacer un análisis filogenético conjunto observan que las cepas se agrupan en 6 filogrupos, los filogrupos A, B1, B2, D y E, previamente descritos por otros autores, y un filogrupo nuevo que denominan F, en donde se encuentran cepas previamente asignadas al grupo D de acuerdo con la PCR triple. Además, Clermont et al. (2011) trabajan con 234 cepas de E. coli aisladas de humanos y animales, en las que detectan la presencia de 7 grupos filogenéticos (A, B1, B2, C, D, E y F); el filogrupo C en la PCR triple se identificaba como filogrupo A. Por ello, Clermont, Christenson, Denamur & Gordon, (2013) hacen una revisión de la PCR triple previamente propuesta y plantean el uso de una PCR cuádruple, donde modifican la secuencia de nucleótidos de los genes chuA, yjaA y TspE4.C2 y adicionan los genes arpA y trpA para confirmar los filogrupos D y C respectivamente. Esta metodología proporciona una correcta asignación en alrededor del 95% de las 234 cepas analizadas. Estos datos se validaron al comparar el filogrupo previamente establecido mediante la secuenciación de los ocho genes de mantenimiento utilizados en el esquema del MLST del Instituto Pasteur, con los resultados obtenidos por la PCR cuádruple. Para el procesamiento de los datos de la secuencia de nucleótidos y ubicar el filogrupo se utilizaron las herramientas BAPS y STRUCTURE, que emplean un algoritmo de asignación poblacional (Clermont et al., 2011; Gordon, Clermont, Tolley & Denamur, 2008).
En la actualidad con el incremento de datos de WGS se han podido determinar con mayor precisión los grupos filogenéticos y con potencial de reportar nuevos filogrupos. Lu et al (2016), mediante la WGS de cepas de E. coli aisladas de Marmota himalayana, identificaron dos nuevos grupos filogenéticos (G y H) que divergen antes que los otros siete filogrupos previamente reportados. De estos dos nuevos grupos, el filogrupo H solo está en una cepa, por lo que aún no hay datos suficientes para considerarlo como un filogrupo nuevo. El filogrupo G se localiza en una colección de cepas previamente reportadas de acuerdo con la PCR cuádruple, como F o B2 demostrando que el filogrupo G es un grupo intermedio entre estos dos (Clermont et al., 2019).
González-Alba, Baquero, Cantón & Galán (2019), trabajan con 6,220 genomas de E. coli disponibles en las bases de datos del NCBI y realizan una reconstrucción estratificada de la diversificación de los filogrupos de la bacteria, a partir de la construcción de un árbol filogenético, ellos reportan que los filogrupos se encuentran en 3 linajes: el EB1A (con los filogrupos E, B1 y A), el D y el FGB2 (filogrupos F, G y B2). Además, mediante el uso de SP-mPH6, observaron que el filogrupo D es el que presenta un menor número de cambios en su genoma, las cepas de este compartían un 99% de los genes ancestrales de E. coli. Del linaje FGB2, el primero en divergir del genoma ancestral hipotético fue el B2, seguido por el G y al final el F. En el linaje EB1A, el A fue el primero en divergir y el E el último. El C, no pudo ser diferenciado del B1. Dado que solo hay una cepa que pertenece al H, no se encontró información suficiente para validarlo como un filogrupo nuevo. Del mismo modo, un grupo de cepas (0.3%) no las pudieron clasificar en alguno de los grupos filogenéticos. Dentro de este 0.3%, 23 genomas (el 0.2% del total de genomas) se agruparon dentro de una rama monofilética que en un futuro podrían considerarse como el filogrupo I. Sin embargo, aún se necesitan más estudios y nuevas herramientas bioinformáticas para proponer la presencia de nuevos filogrupos.
Por otro lado, Abram et al. (2021) mediante un análisis basado en Mash7 de 10,667 genomas ensamblados y 95,525 genomas no ensamblados de E. coli, determinaron la presencia de 12 filogrupos (A, B1, B2-1, B2-2, C, D1, D2, D3, E1, E2 (O157), F y G). Estos comparten genes centrales únicos entre cada uno de ellos que no están en el genoma central de los demás. También observaron que el genoma central de los filogrupos denominados como patógenos (B2-1, B2-2, D1, D2, D3, E2 (O157), F y G) están más conservados que los identificados como cepas comensales (B1 y A), lo que concuerda con trabajos previos donde los eventos de recombinación son menos frecuentes en cepas de E. coli patógenas (intra y extraintestinales) que en cepas comensales (McNally, Cheng, Harris & Corander, 2013). Se vieron discrepancias en la clasificación del 7% de los genomas entre el análisis basado en Mash y la PCR del ClermonTyping8. Otro 9% de ellos no se pudo clasificar mediante esta herramienta y 47 más del filogrupo G quedaron como F con base en ClermonTyping.
Los trabajos de González-Alba et al. (2019) y Abram et al. (2021) indican que el uso de herramientas analíticas en los genomas secuenciados permite la determinación de cambios genómicos entre los diversos filogrupos, lo cual es útil para el entendimiento de la diversidad genómica de microorganismos como E. coli (Figura 4).
Secuenciación del genoma completo (WGS) de E. coli uropatógena (UPEC)
La WSG ha revolucionado diversas áreas de la investigación, incluida la microbiología. Desde la aparición de los primeros genomas bacterianos completos, se han secuenciado miles incluyendo cepas de referencia y aislados de diversos orígenes. E. coli es uno de los microorganismos más estudiados, como también se mencionó, en el laboratorio a nivel mundial, en la actualidad en la base de datos del GenBank se encuentran un total de 255,927 genomas obtenidos de diversos nichos ecológicos. Estos se encuentran hasta febrero del año 2024, en diferentes niveles de ensamble, siendo los no cerrados (incompletos: scaffold y contig) los más predominantes (Figura 5) (Genome-NCBI-NLM, n.d.).
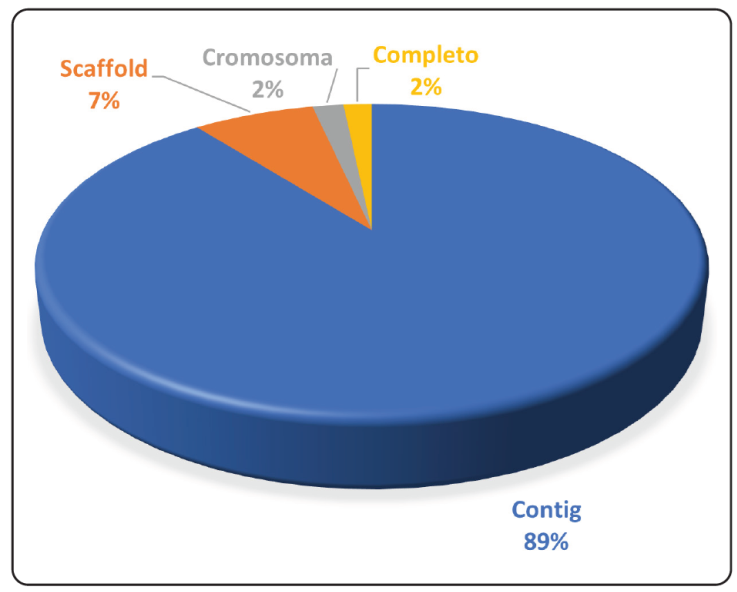
Creación propia realizada en Excel.
Figura 5 Nivel de ensamble de los 255,927 genomas depositados en la base de datos del NCBI. Desde el año 1980 a febrero de 2024 se han depositado diversos genomas de libre acceso. Contig: genomas no cerrados (incompletos); Scaffold: genomas no cerrados fragmentados compuesto por contigs y gaps (incompletos); Cromosoma: Genoma cromosómico cerrado (completo); Genoma completo: Genoma cromosómico y plasmídico (sí los contiene) (Genome-NCBI-NLM, n.d.).
La disponibilidad de estos genomas ha permitido la identificación de diversos genes relacionados con la virulencia, la formación de biopelículas y con la resistencia a antibióticos incluidos los presentes en islas genómicas (GEIs, por sus siglas en inglés), (Subhadra et al., 2018; Torres-Puig et al., 2022; Welch et al., 2002). El libre acceso a genomas secuenciados ha permitido la creación de la Base de Datos De Factores De Virulencia (VFDB) y la Base de Datos Integral de Resistencia a los Antibióticos (CARD). Estas bases sirven como servidores en línea para la identificación de genes de virulencia y de resistencia respectivamente, así como de mutaciones en sitios blanco de antibióticos que confieren resistencia (Alcock et al., 2023; Liu, Zheng, Jin, Chen & Yang, 2019). Son también utilizadas como base por otras plataformas en línea como PATRIC, VirulenceFinder y VFanalyzer para factores de virulencia y ResFinder y PointFinder para resistencia y así como ABRricate para ambos. Estas plataformas realizan una búsqueda mediante BLAST para la determinación in silico de los factores de virulencia y resistencia en genomas secuenciados, la mayoría, están en constante actualización (Bortolaia et al., 2020; Joensen et al., 2014; Seeman, 2016; Wattam et al., 2017).
La disponibilidad de los genomas en conjunto con las bases de datos ha permitido el desarrollo de trabajos bioinformáticos integrales sobre la diseminación de resistencia entre cepas clínicas, así como las discrepancias entre lo reportado fenotípicamente con lo reportado a nivel genómico. Por ejemplo, en el diagnóstico de susceptibilidad a la amoxicilina con ácido clavulánico, por una concentración estandarizada para pruebas de susceptibilidad antibiótica, puede dar lugar a falsas cepas sensibles (Sánchez-Osuna, Barbé, & Erill, 2023; Vanstokstraeten et al, 2023). Por ello, la WSG no solo ha proporcionado un mayor entendimiento en la diseminación de resistencia, sino que puede servir como un valioso apoyo para la predicción de la resistencia fenotípica de diversas cepas mediante el uso de más de una herramienta bioinformática.
Las cepas de UPEC pueden ser parte asintomática de la microbiota intestinal. Sin embargo, una vez que UPEC accede al tracto urinario causa ITU que puede ser recurrente, crónica o en casos graves progresar a una urosepsis (Desvaux, Dalmasso, Beyrouthy, Barnich, Delmas & Bonnet, 2020). Si bien la serotipificación, la filogenia y la MLST han servido en la detección de brotes, para la vigilancia epidemiológica, el conocimiento que se tenía sobre UPEC se encontraba restringido a genes conservados (Figura 3). Las regiones de estos genes conservados han proporcionado escasa información sobre su evolución como patógeno, ya que no hay datos sobre los procesos por los cuales los factores asociados a la patogenicidad han sido adquiridos por UPEC con el paso del tiempo (Lo, Zhang, Foxman & Zöllner, 2015).
La información sobre la evolución patogénica de UPEC ha sido proporcionada por la secuenciación del genoma completo (WGS) de cepas clínicas que han sido usadas como modelos de estudio para el entendimiento de su mecanismo de patogenicidad y el origen de los genes asociados a este evento.
El genoma de las cepas prototipo de UPEC oscila entre 5.23 (CFT073), 5.07 (UTI 89) y 4.93 (536) Mpb y es mayor al tamaño del genoma de una cepa comensal como el de E. coli K-12 substr. MG1655 (4.64 Mpb). Se estima que la diferencia en tamaño entre una cepa patógena y la comensal puede llegar a ser hasta de 1 Mpb. La diferencia de tamaño se asocia con la ganancia y pérdida de los genes relacionados a factores de virulencia y/o resistencia a través de elementos genéticos móviles como las secuencias de inserción, los transposones, los plásmidos y los bacteriófagos diseminados mediante eventos de transferencia genética horizontal (TGH) como la transformación, la transducción y la conjugación bacteriana (Croxen, Law, Scholz, Keeney, Wlodarska & Finlay, 2013; Croxen & Finlay, 2009).
Se requiere una mejor comprensión sobre los genes que codifican para los factores de virulencia y de resistencia clínicamente relevantes, su entorno genético, su regulación y como fueron adquiridos para el desarrollo de nuevas estrategias terapéuticas y profilácticas que puedan contribuir en la reducción de la morbilidad asociada con las infecciones urinarias causadas por UPEC.
Los avances en WSG junto con el análisis bioinformático, para la interpretación de los datos de la secuencia genómica, han respondido a cuestiones básicas de la evolución del genoma de E. coli y específicamente de UPEC que la ha llevado a adaptarse mejor al tracto urinario (Figura 6). Además, los estudios de genómica comparativa mediante el uso de diversas herramientas bioinformáticas y analíticas de las cepas de UPEC sugieren que su evolución a partir de una cepa comensal se debe a la adquisición de GEIs mediante diversos eventos de transferencia genética horizontal. Se ha demostrado que las GEIs en UPEC contribuyen a la aptitud y a la adaptación bacteriana a nuevos nichos ecológicos dado que portan genes relacionados a la virulencia (adhesinas, toxinas, formación de biofilm) y resistencia antibiótica (beta-lactamasas), (Desvaux et al., 2020, Dobrindt, Hochhut, Hentschel & Hacker, 2004; Tenaillon et al., 2010).
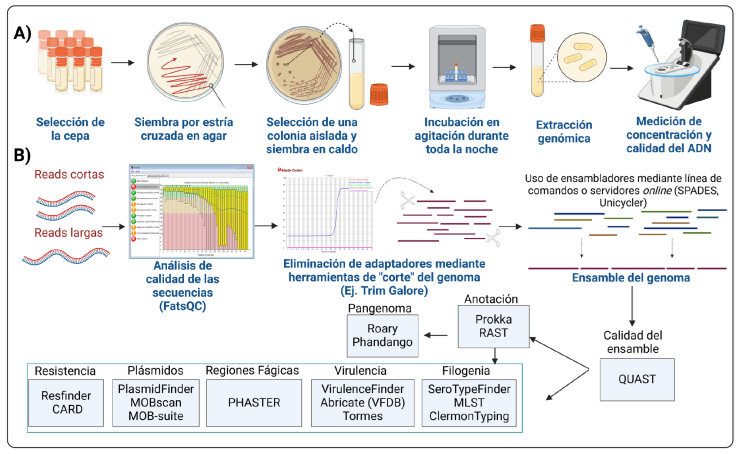
Creación propia realizada en BioRender.com
Figura 6 Guía para la secuenciación del genoma completo. A) Pasos previos a la secuenciación del genoma. Realizar una buena extracción de ADN de una colonia aislada de la cepa bacteriana problema. Se requiere que el ADN tenga una concentración > 3µg de ADN genómico y una relación de absorbancia de 260/280 de 1.8 a 2.0 y absorbancia de 260/230 de 2 a 2.2 B) Análisis de calidad, ensamble y análisis bioinformático. La calidad de las secuencias se determina usando la herramienta FastQC que da una distribución idónea de las puntuaciones de calidad de las secuencias (Por ejemplo: calidad de secuencia por base, el contenido de secuencia por base y la presencia de adaptadores), la presencia de adaptadores interfiere al realizar el ensamble. Continúa con la eliminación de adaptadores, ensamble, determinación de la calidad del ensamble. Finalmente, el análisis bioinformático incluye el uso de varias herramientas.
El análisis del genoma de la cepa prototipo de UPEC CFT073 aislada de una mujer con pielonefritis aguda en EE. UU., muestra que las partes conservadas de su genoma o “core genome” han sufrido cambios lentos en su estructura. Sin embargo, en el resto de su genoma o “genoma accesorio” se presentan una serie de pérdidas y ganancias de genes recién introducidos a través de numerosos eventos de TGH. Algunos de los sitios usados en la TGH sirven como objetivos de inserción universal o “sitios calientes” utilizados de forma independiente en linajes separados (Luo, Hu, & Zhu, 2009; Welch et al., 2002). Además, al hacer una comparación entre cepas de UPEC con cepas de E. coli comensales se observa que las cepas de UPEC son oportunistas y no muestran una selección preferencial por los factores de virulencia que adquieren. Como si no fuera suficiente, las cepas de UPEC que se encuentran estrechamente relacionadas a nivel filogenético pueden tener fenotipos uropatógenos diferentes (Lo et al., 2015).
La cepa UPEC 26-1 fue aislada de un paciente que sufría de pielonefritis aguda en Corea, con una alta virulencia y al comparar su genoma se encontró que contiene 98,325 pb adicionales al de CFT073, donde se encuentran genes que codifican para proteínas involucradas en la virulencia (por ejemplo, adhesinas), resistencia a múltiples fármacos (bomba de expulsión MFS resistente a tetraciclina) y formación de biopelículas (sistema toxina-antitoxina), (Subhadra et al., 2018).
Además, la WGS ha permitido dilucidar la carga genética de las cepas previamente utilizadas como modelos en pruebas fenotípicas, como CI5 y NU14 cuyo genoma ha servido para el entendimiento del ciclo infectivo de UPEC, así como su capacidad para formar colonias bacterianas intracelulares. Estas cepas han servido también de modelo para el estudio de vacunas usadas en el tratamiento de las ITU por UPEC (Mehershahi, Abraham & Chen, 2015; Mehershahi & Chen, 2017).
En México aun cuando se cuenta con poca información sobre el genoma completo de las cepas de UPEC que circulan dentro de nuestra población, en el año 2019, Paniagua-Contreras et al. (2019) realizaron la WGS de 24 UPEC aisladas de pacientes femeninos (n=20) y de pacientes masculinos (n=4) y observaron que las secuenciadas presentan un genotipo de resistencia a múltiples fármacos de uso común en la clínica. Además tienen una gran cantidad de genes de virulencia, su pangenoma es “abierto” y con una gran familia de genes ortólogos9 (Paniagua-Contreras et al., 2019).
Conclusiones
Las infecciones del tracto urinario son de las enfermedades más recurrentes a nivel mundial que afectan tanto a hombres como a mujeres, siendo éstas últimas las más propensas a padecer la enfermedad, pero en los varones es más grave. E. coli uropatógena es el principal agente etiológico de las ITU que provoca más del 70% de estas infecciones. Las nuevas tecnologías de secuenciación del genoma completo se han convertido en una herramienta poderosa para describir la relación que existe entre la presencia de algunos genes, principalmente aquellos relacionados a su clasificación filogenética, virulencia y resistencia bacteriana, con fenotipos que le han permitido a este importante patógeno diseminarse mundialmente.
El número de secuencias del genoma completo de E. coli disponibles en las bases de datos de acceso libre al crecer de forma exponencial igual que las herramientas bioinformáticas permiten realizar estudios integrales de genómica comparativa donde se emplean diversas herramientas. Estos trabajos son de gran valor, no sólo, para entender el potencial de virulencia de UPEC, sino también para clasificarla filogenéticamente con la ayuda de “biomarcadores específicos” compartidos por todas las cepas UPEC obtenidos del análisis de WGS de diversas cepas alrededor del mundo. Sin embargo, esto no descarta la posibilidad de hallar genes que sean diferentes de acuerdo con la región geográfica de donde estas procedan. Así como continuar con el estudio de los genomas de E. coli, en especial del patotipo uropatógeno de la población mexicana aislado de ITU, para un mejor control y tratamiento de la enfermedad.

