











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Deoxyribonucleic acid, commonly known as DNA, is a large and complex molecule located in the nucleus of cells in all living organisms. It takes the shape of a double helix structure, where the genetic information unique to each individual and species is stored. All life is based on a code written in DNA molecule, the common denominator among living beings [1]. DNA can exist in a single-stranded form or as higher-order structures, including the canonical double helix and noncanonical duplex, triplex, and quadruplex species [2]. In a DNA molecule, each sugar in the two strands is attached to one of four nitrogenous bases - Adenine (A), Guanine (G), Cytosine (C), and Thymine (T). These bases can bond through hydrogen bonds, and their bonding possibilities differ. Specifically, DNA's double helix structure is attributed to hydrogen bonds between A-T (two) and C-G (three) base pairs[3].
The number of hydrogen bonds determines the specific pairing between nitrogenous bases, but this structure is prone to errors and can sometimes be repaired [4] [5].
According to the NCBI database [6], the number of records has doubled approximately every 18 months since 1982[7]. From a computer science perspective, advanced metaheuristic strategies are required to handle such information[8] [9].
We developed a genetic algorithm to perform multiple genetic sequence alignments in this work. The objective is to explore the improvement from different parametrization schemas (Gp1, Gp2, Gp3, and Gp4) for the algorithm`s performance in terms of a) the solution quality (fitness) achieved by the algorithm and b) the computational effort (
In the field of genetics, the parallelization strategy of processing information has shown significant improvement in the results, especially in reducing the computational time required to solve various problems[10] [11] [12]. As a validation method, the numerical performance profile measures the relative efficiency of the algorithm in solving the set of problems[13].
Bioinformatics software testing is often complex due to the difficulty in verifying the correctness of output and effectively generating failure-revealing test cases[14]. In this work, we developed four strategies based on a genetic algorithm for the multiple sequence alignment problem and compared their performance to each other. Multiple sequence alignment (MSA) is essential in bioinformatics, aiding in phylogenetic, protein, and genomic analysis, but faces challenges in increasing alignment accuracy[15].
There are several algorithm strategies in the state of the art, such as the "Progressive alignment," which has been one of the most used methods since the 1980s [16]. In addition to improvements in algorithm design, modern hardware technology has increased processing speed and enabled more simultaneous alignment sequences[17]. Furthermore, integrations based on framework processing like Apache Spark can produce promising results and new perspectives [18].
Regarding the processing algorithms, researchers have developed exact, progressive, and iterative algorithms to address the Multiple Sequence Alignment (MSA) problem. Exact algorithms exhaustively search in the space of solutions and calculate the global optimum in sequences of relatively small lengths. However, they cannot guarantee finding the best solution in arrangements of hundreds or thousands of bases. Blast is an example of this type of algorithm [19]. On the other hand, progressive algorithms work by iteratively improving a sequence alignment by adding new sequences. The process involves gradually building upon an initial alignment until an optimal solution is found [20] [21]. Dynamic programming algorithms are commonly used to implement strategies that explore solution spaces using a substitution matrix [22]. One example is the ClustalW algorithm [23]. However, errors at the beginning of an alignment can propagate to the rest of the operation, which is a drawback of these algorithms. Furthermore, another strategy to tackle bigger MSA problems is to iteratively perform partial alignments [24] [25].
On the other hand, Genetic algorithms are iterative metaheuristic strategies based on bioinspiration. A thorough analysis of the algorithms used in this context can be found in [26]. Due to the metaheuristic origin of Genetic Algorithms, which does not guarantee achieving the optimal solution, we assume no comparison between metaheuristics and/or stochastic strategies vs. progressive or deterministic strategies. However, metaheuristics permit working with more extensive problem instances.
This work presents a bioinformatics breakthrough and builds on fundamentals established by previous papers, such as [27], highlighting the importance of hardware acceleration in DNA sequence alignment. The utility of parallel processing was established by [28], which improves the process at the bit level. This work presents an innovative methodology that combines multiple techniques, including genetic algorithms, to enhance multiple sequence alignment. The methodology also employs the algorithms' performance profile as an efficient comparison method.
Due to the increasing scientific research in medicine-based informatics, the genetic algorithm has been tested using a set of genetic sequences from a group of vegetables of particular interest in medicine, such as Euphorbia pulcherrima, which has Anti-Inflammatory, Analgesic, Sedative, and Muscle-Relaxant Activities [29]. Preventing health complications is a significant motivation for studying the properties of vegetables.
Materials and methods
The computational processes in this study were meticulously developed on four identical personal computers (PCs) with a Microsoft Windows operating system. These PCs were equipped with 8 GB of RAM and an Intel core i5 processor, ensuring the study's robustness and reliability. The algorithms were programmed in Java with Netbeans IDE, a widely recognized and trusted platform for software development.
Genetic Algorithm
In this study, a genetic algorithm (GA) is used to simulate the natural adaptation process of living beings. This computer-based representation was initially proposed by [30]. These are some instances of how GA’s are used:[31] [32] [33] [34]. While most search algorithms operate on a single solution, a GA operates on a population of solutions. The classical GA codes a given problem, generating a population of potential solutions. This method involves crossing, leading to mutation, and discovering new and improved solutions. This statement is founded on Darwin's evolutionary theory.
Sequence Alignmen
When there are two sequences (m,n) that can accommodate a limited number of gap insertions, the number of alignments increases as the length of the sequences increases. The number of potential alignments for a pair of sequences m and n, with 'k' insertions, can be calculated using Equation (1) as stated by Waterman in 1995:
When attempting to solve the multiple sequence alignment problem using brute force, the problem becomes NP-complete. In contrast, dynamic programming has a complexity of O(LN), where L is sequence length, and N is the number of sequences. Researchers aim to enhance MSA efficiency via heuristic and metaheuristic approaches and parallel implementations to reduce computational costs [35].
Sum-of-pairs multiple alignments: The given strings, represented by S1 to Sk, are used to create multiple alignment. This alignment consists of strings T1...TK, which are the same length as the original strings. The new strings are made by inserting spacing symbols at specific positions in the original strings. However, only columns full of spacing symbols are not allowed.
Given a multiple alignment T1...TK of length m and a scoring function σ for pairs of characters (σ(a,b)) and pairs of character and spacing symbol (σ(a,-) or σ(-,b)), we define its sum-of-pairs score as described in Equation (2):
Compute a multiple alignment of strings S1 to Sk with a given scoring function sigma to find the maximum sum-ofpairs score.
Problem Codification: Text or binary sequences are commonly used for multiple sequence alignment. These sequences include array-like data structures, making handling characters and reordering positions easier. As a result, the computational effort required by the processing machine is reduced. In this work, each alignment matrix A represents an individual, and each matrix position contains a single character (a). Equation (3) shows the matrix positions where a nucleotide or a gap can be found.
Each row in the alignment represents a sequence of the set to be aligned.
Proposed genetic algorithm: GAAPd
The algorithm starts with an Initial Population (Pi) of 500 individuals, which is later reduced to a fixed number of 100 individuals (Pt) between 100 generations. This helps maintain stability in the algorithm and prevents excessive computational resource usage. The individuals in the initial population are generated by random potential solutions to the problem.
Individuals are evaluated using a parallel process and the widely used Blosum evaluation matrix [36]. This matrix helps measure the quality of the in-turn alignment. There are ongoing efforts to parallelize genetic algorithms either partially or entirely, and these efforts have demonstrated meaningful results in their respective fields of application [37] [38] [39] [40].
To speed up the evaluation process, we divide the workload and assign it to each evaluator instance, which is a component of the evaluation class designed for this process. We avoid double evaluations of union regions to ensure synchronicity among evaluators.
After completing the task, evaluators assign each population member a fitness value (ft) used on their partial results. Figure 1 illustrates the evaluation process described in this work.
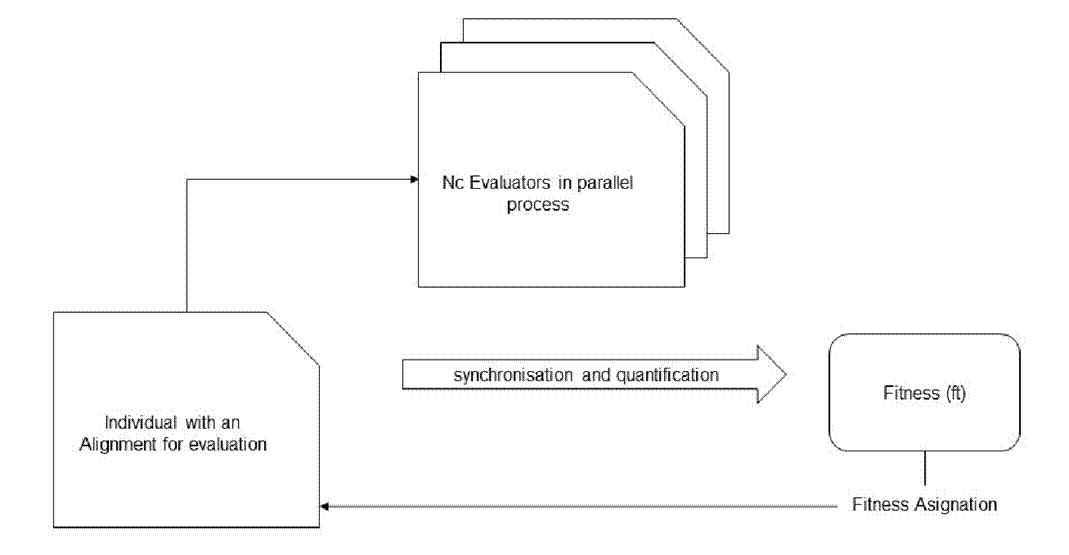
Figure 1 The process of evaluation. Nc refers to the number of evaluator instances created to evaluate everyone. Once the fitness value is assigned, the process is repeated for the following individuals in the population.
According to theory, there is a direct correlation between the number of cores and the reduction of processing time (
Where
During the crossover process, individuals are randomly selected from the best performers of the evaluation process in the Pt set. In this context, it implies that only the most outstanding individuals proceed to the crossover phase. Their probability of being assigned to another individual is Pb= 1/Pt, equal for all.
A random double-point cut is made (CPA,CPB), and each crossover produces two children integrated into the population. The positions of CPA and CPB in the alignment matrix are also random.
After making the necessary cuts for both individuals, three parts of the father (P1F,P2F,and P3F) and three of the mothers (P1M,P2M,and P3M) are obtained.
Child 1 and Child 2 incorporate all six elements shown in Figure 2.
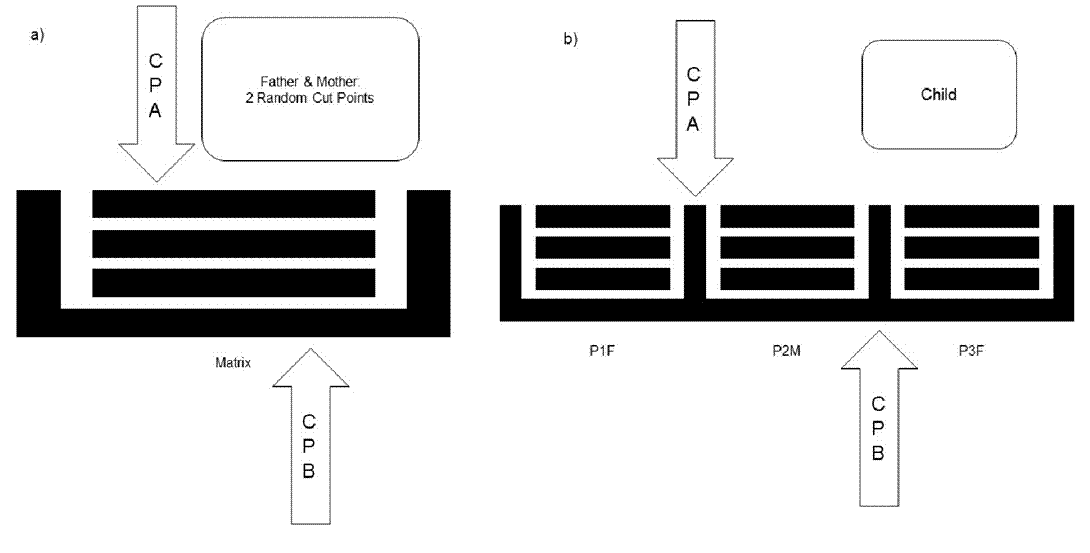
Figure 2 The crossover processes. In this process, two individuals produce three parts each. The resulting child has one part from the mother (P2M) and two from the father (P1F,P3F). Another child has one part from the mother and one from the father.
The mutation process occurs for progenies [41], which is achieved by inserting or deleting gaps using the hyphen character "-" in various positions within the matrix. This process helps to improve the alignment further. The probability of an individual moving to the mutation process is Pm = 0.2, in which changes are randomly applied to the alignment matrix based on the Pm value.
Various mutation techniques have been documented in the literature, each relying on a specific mutation probability [41] [42] [43]. We developed four forms of mutation described as follows:
One of the sequences is randomly selected for mutation, where a maximum of 200 gaps can be inserted at the beginning.
A random point in the matrix is selected for gap insertion. The number of spaces added is randomly determined, up to 300.
Insertion of a gap column at a random point. A gap column is inserted at some position in the alignment matrix.
Random gaps are deleted. Up to 100 gaps are removed, and the number of gaps removed is randomly determined within a range of 1 to 100.
Algorithm Pseudocode and GitHub Access
The pseudocode of the proposed algorithm is presented as follows. Let Nc be the number of processing cores and Nm the mutation level as a repeating mutation process over a given matrix.
1) Population Initialisation (P)
2) Population Evaluation (Blosum Matrix)
a. Distribute partial work for cores (Nc) and evaluator Instances.
b. Execute parallel evaluation process.
c. Once finished, evaluation instances compute the overall fitness value.
3) Worst Individuals Elimination.
4) Crossover.
5) Repeat progeny mutation (Nm) Times.
6) Integrate progeny into P.
The GitHub project is available for further analysis at https://github.com/riosew/GAAPd-Software.git.
The algorithm is presented as a set of corresponding classes. The different versions designated as Gp1, Gp2, Gp3, and Gp4 can be produced by changing the number of cores (Nc) for the parallel process and the mutation level (Nm) to change the mutation cycles. Thus, it can be executed with the different Nc and Nm values in Table 1. It initiates the process with a basic visual interface, where the user can change the indicated values.
Time Complexity Analysis Big O
The evaluation process is divided into the following parts.
Creation of evaluators: This loop runs several times equal to the number of available cores. Inside this loop, another loop runs several times equal to the size of the alignment matrix. Therefore, this part of the algorithm has a complexity of O(n*m), where n is the number of cores and m is the size of the matrix.
Waiting for all evaluators: This loop runs until all evaluators have finished. In the worst case, this could take time proportional to the evaluator, which takes the longest to complete the work.
Traversal of evaluators adding results: This loop runs several times equal to the number of evaluators (equal to the number of cores). Therefore, this part of the algorithm has a complexity of O(n), and the total complexity of the evaluation process is O(n*m), where n is the number of cores and m is the size of the matrix. However, the complexity analysis of the nested cycles in the micro evaluator instance (mE) is worth noticing. The mE instances are as many instances as the core Nc value. The complexity of the mE process is O(nmp2). Since n and m are the matrix size, and p is the vector size in the Blosum evaluation matrix, which performs a traversing process for n and m. Finally, in big(O) analysis, the worst case of operation establishes the total algorithm complexity.
Experimental Method
This paper's experiment describes a sequence of algorithms and the Performance Profile.
Set of Sequences
The dataset has been expanded by integrating 13 sequence groups, described in Table 2. It is available for future reference on the Galaxy platform [44] at https://usegalaxy.org/u/rioswillars/h/dataset-for-genetic-algorithm. Each group comprises N sequences of varying lengths.
Table 2 Test Sequence Sets.
| Set | Name | N |
|---|---|---|
| 1 2 3 |
Agave victoriae reginae Aristida purpurea Bellucia grossularioides |
38 38 38 |
| 4 5 6 7 8 9 10 11 12 13 |
Bursera simaruba Cnidoscolus urens Cordia alliodora Curatella Americana Cydista diversifolia Echinocactus platyacanthus Ephiphylum hookeri Erythrina herbacea Euphorbia pulcherrima Gossypium arboretum |
35 35 28 33 29 38 31 35 23 29 |
Algorithms
Four versions of the GAAPd algorithm were created to evaluate the number of evaluated functions (
Determination of numerical performance profiles
A comparative analysis was conducted to determine the robustness and efficiency of four methods: Gp1, Gp2, Gp3, and Gp4. The study used numerical performance profiles, which define the cumulative distribution function for a numerical performance profile. The metrics of interest were the algorithm's execution time, its ability to reach the global optimum in the objective function, and the number of evaluations performed during the calculation sequence. These factors were compared to analyze the performance and convergence of the optimization method.
This paper aims to compare four different methods based on three metrics. The first metric is the relative distance between the optimal solution found by the optimization method and the known global optimal solution obtained by the ClustalW algorithm. This metric measures the robustness of the process, i.e., its ability to locate the global optimum of the objective function. The second and third metrics are the number of function evaluations
To evaluate specific metrics, we considered four optimization methods (ns = 4) and 13 problems or sets of sequences (np = 13). Each case study was solved 30 times, with random initial solutions and different random alignments. The stop condition was stabilized within the 100-generation limit. However, since the stop criterion is based on the generation number, a sensitive tolerance of 1.0E-06 is assumed for the value of the objective function since the optimal solution is uncertain. It was set as the convergence criterion for the four methods for all four optimization methods and sequence sets, the values of tp,s were calculated using the results of the 30 calculations and the following expressions (Equation 5).
Being
Where S corresponds to the set of optimization methods analyzed. This method assigns a value of 1 to the algorithm that performs the best in each problem. Finally, the cumulative probability rate ρs(τ) for the optimization strategy S and the metric in question is defined as in the Equation (7):
Where τ is a factor that is defined in (1, ∞). In the graph of the performance profile, for example, the graph of ρsversus τ, compares the relative performance between optimization methods for the problem group. So far, performance profiles have been used by [46] to compare bioinspired algorithms and benchmark functions. However, this concept has not been used to compare the methods described in the MSA problem.
Results and discussion
The results of the fitness,
Table 3 Mean values for the Fitness metric of the Gp1, Gp2, Gp3 and Gp4 algorithms for the 13 multiple sequence alignment problems.
| C | Gp1 | Gp2 | Gp3 | Gp4 |
|---|---|---|---|---|
| 1 | 62723.7 | 62385.6 | 63085.2 | 63105.4 |
| 2 | 59747.1 | 60072.8 | 59776.5 | 59605.0 |
| 3 | 57785.0 | 58536.2 | 58413.7 | 57109.7 |
| 4 | 52653.2 | 53250.0 | 53497.3 | 53526.8 |
| 5 | 16934.0 | 16463.8 | 16917.7 | 17270.7 |
| 6 | -4932.2 | -4724.0 | -5529.0 | -4903.8 |
| 7 | -43993.1 | -44014.3 | -43848.8 | -43909.3 |
| 8 | -33991.1 | -33350.3 | -33782.7 | -33775.6 |
| 9 | 54515.2 | 54405.9 | 54311.7 | 53919.9 |
| 10 | -99067.2 | -98881.0 | -98633.5 | -98737.2 |
| 11 | 52933.6 | 53749.8 | 52805.7 | 52540.1 |
| 12 | -4082.1 | -4463.7 | -4449.5 | -4134.2 |
| 13 | 9190.8 | 8423.8 | 8997.1 | 8699.8 |
The fitness metric shows promising performance results for the Gp2 and Gp3 algorithms since the objective is to increment the most possible fitness value. Also, the negative values describe complicated sequences for the alignment problem for all four algorithms.
For the number of evaluated functions (
Table 4 The
| C | Gp1 | Gp2 | Gp3 | Gp4 |
|---|---|---|---|---|
| 1 | 690.867 | 684.067 | 679.600 | 688.733 |
| 2 | 680.400 | 693.033 | 686.433 | 685.100 |
| 3 | 684.333 | 690.700 | 686.233 | 686.600 |
| 4 | 686.033 | 693.167 | 688.867 | 687.900 |
| 5 | 685.300 | 687.167 | 683.233 | 681.033 |
| 6 | 684.667 | 689.867 | 683.633 | 684.167 |
| 7 | 692.133 | 683.167 | 690.233 | 693.933 |
| 8 | 682.367 | 690.400 | 679.600 | 689.867 |
| 9 | 686.200 | 689.400 | 684.533 | 681.067 |
| 10 | 693.400 | 685.667 | 686.733 | 686.733 |
| 11 | 685.233 | 680.767 | 693.333 | 690.700 |
| 12 | 682.733 | 683.367 | 683.867 | 683.100 |
| 13 | 685.933 | 681.500 | 683.967 | 683.600 |
The time it takes for the algorithms to process the stated problems is a commonly used metric for comparing the strategies' performance. Table 5 shows the performance in terms of the required time for each of the four algorithms to process the 13 multiple alignment problems. It is worth noting that Gp3 took more time than the other algorithms to process the alignment problems. Furthermore, Gp4 showed better performance than the different algorithms. The efficiency of an algorithm is not determined by the time it takes to run. Still, in this case, the time comparison helps show the main difference between the algorithms: the number of nuclei for the hardware process.
Table 5 The comparative of the mean values in the metric Time for the four algorithms in the 13 sets of sequences for the multiple alignment problem.
| C | Gp1 | Gp2 | Gp3 | Gp4 |
|---|---|---|---|---|
| 1 | 4409.033 | 3943.900 | 5065.433 | 3381.000 |
| 2 | 4118.767 | 3554.400 | 4849.733 | 2964.267 |
| 3 | 4582.700 | 3944.633 | 5352.100 | 3268.467 |
| 4 | 3779.267 | 3289.667 | 4397.400 | 2727.467 |
| 5 | 3542.733 | 3141.700 | 4128.300 | 2598.000 |
| 6 | 2649.367 | 2543.267 | 3106.400 | 2061.000 |
| 7 | 3439.833 | 3138.667 | 4025.133 | 2667.833 |
| 8 | 2929.867 | 2753.867 | 3396.033 | 2300.000 |
| 9 | 4546.700 | 3874.767 | 5232.233 | 3361.800 |
| 10 | 5006.133 | 4514.933 | 5748.400 | 3919.100 |
| 11 | 3866.033 | 3288.933 | 4525.900 | 2901.100 |
| 12 | 2501.500 | 2321.367 | 2912.000 | 2032.300 |
| 13 | 4440.800 | 3894.400 | 5086.733 | 3478.067 |
We present a Microsoft Excel file named results in the provided github link for a detailed description of the tables above.
Figure 3 displays the performance profile for the metric
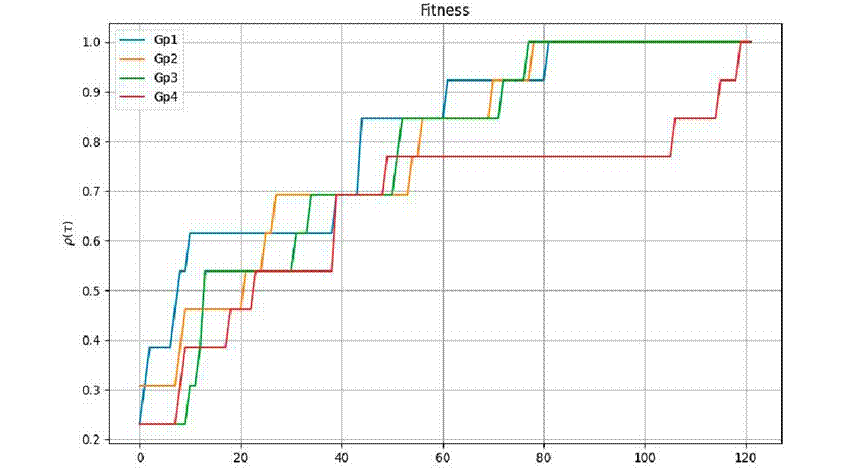
Figure 3 Numerical performance profile for fitness metric
Regarding efficiency, the metric
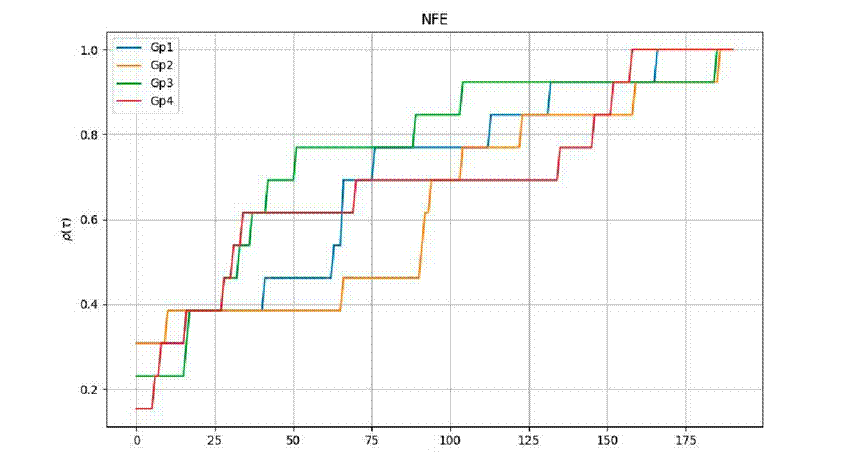
Figure 5 indicates that the Gp4 optimization method outperforms the others in terms of efficiency, as measured by the time spent on optimization
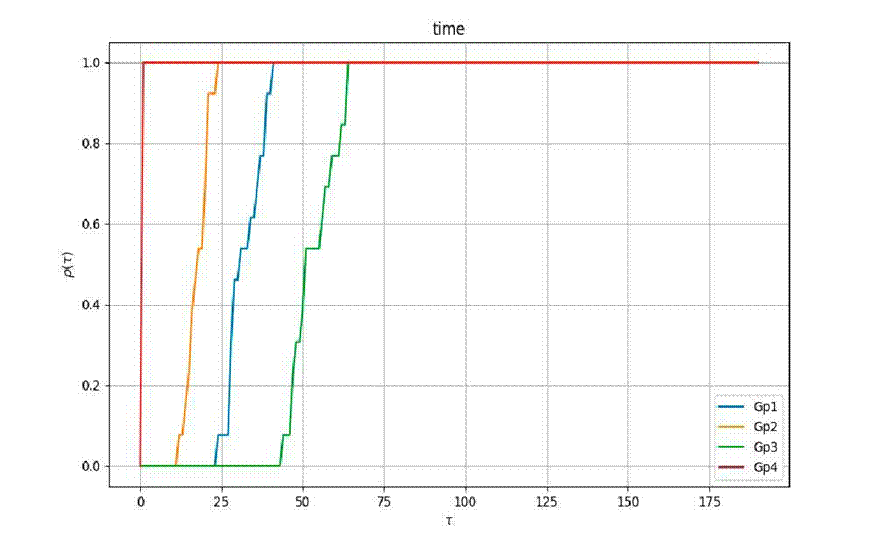
However, the Gp4 algorithm did not produce remarkable fitness or
Conclusions
This paper compares and discusses four strategies of the Genetic Algorithm method using the numerical performance profile model [47] in a set of genetic sequences. Based on the findings, the Gp1 approach is the most robust compared to other methods studied in this research. However, the efficiency, measured by the number of functions evaluated and convergence time, varies between the alignment methods. Gp4 is the method with the fewest evaluated functions and the highest processing speed. Thus, it can be concluded that the number of cores influences the alignment method's efficiency in parallel processing, and the robustness of the alignment method is related to the number of mutations. However, the Gp2 algorithm, with the highest core number and mutation level, did not perform remarkably well compared to the other strategies that varied the mutation level and number of cores.
Increasing the sequences in the data set is recommended to obtain more accurate results. By testing with the data set used in this research, these findings can be compared to other studies on genetic algorithms. Finally, it is essential to note that variations in mutation levels or other parameters can affect the performance of the different strategies.
On the other hand, conventional computer equipment can produce promising results when processing genetic information if the algorithms are specifically designed to make the most of hardware resources. Additionally, the computational effort of the hardware used impacts the number of evaluated functions. The quality of the solution obtained in the case of multiple sequence alignment relies on specific parameters of the genetic algorithm, such as the size of the population, the mutation level, and the crossing method. Moreover, the comparison method based on the determination of the performance profile is recommended as a valuable strategy for contrasting results in different metrics of interest.

