











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Because of increasing life expectancy in the population, there is a growing number of persons reaching old age and associated comorbidities. One of the most frequent observed morbidity in the elderly is heart failure, and at least fifty percent of these patients have the variety of heart failure with preserved ejection fraction (HFPEF). Usually the variety of HFPEF is related to their old age and/or associated with comorbidities such diabetes mellitus, systemic hypertension or ischemic heart disease, or simply inappropriately categorized by not appropriately studied. Cardiac amyloidosis is usually considered a rare disease and an uncommon cause of heart failure in the elderly. It is very probable that cardiac amyloidosis is underdiagnosed and more prevalent than usually thought in the elderly, especially in those with HFPEF.1-3 Amyloid was described in 1858 by Rudolf Virchow when he observed tissue deposits of a «starch-like» material or amyloid.4 Lately, with new diagnostic methods there has been increased interest in cardiac amyloidosis, and as consequence it now seems it is not so infrequent as previously thought.5,6 If cardiac senile amyloidosis is the etiology in 10 or 15% of patients with HFPEF, disease for whom there is no clearcut therapy, a noninvasive diagnosis may make it easier to recruit patients for clinical trials that are heavily needed to treat this commonly underdiagnosed and suboptimal treated disease. When we refer to cardiac senile amyloidosis we mean amyloid heart disease related to the protein precursor wild-type transthyretin (wild-type TTR), it is necessary mention briefly other types of abnormal TTR such as variant-mutant forms of TTR and light chain AL amyloidosis that can express also in the elderly.
Epidemiology
Heart failure in the elderly is not uncommon, and HFPEF is responsible for at least 50% of these cases. Clearly, not all the elderly patients with heart failure have one of the common systemic disease such as diabetes mellitus, systemic hypertension or ischemic heart disease related to this variety of heart failure. Autopsy studies in the elderly have disclosed amyloid deposits in 25% of the hearts in octogenarians.7,8 Also, there has been reports that at least 13% of heart failure in the elder might be related to amyloid infiltration. Unfortunately, for several reasons, there is under diagnosis of amyloid heart disease in the older population, and as consequence suboptimal knowledge of its pathophysiology and treatment.9 In the United States 3,000 new cases of AL amyloidosis are diagnosed each year, 30 to 50% with cardiac involvement and 10 to 15% associated with multiple myeloma.10 The population with ATTR cardiac amyloidosis is not so well known, but recent data suggest it is not as infrequent as previously thought, probably it is underdiagnosed, and it may be responsible for several cardiovascular disease expressing in older people such as HFPEF, low-flow aortic stenosis, and atrial fibrillation.11,12
Pathophysiology
Amyloid consist of an abnormal and insoluble misfolded beta-pleated protein that deposits in the interstisium of different tissues, disrupting the structure and function of interstitial space and adjacent cells. There are several different amyloid protein precursors and at least 30 precursors have been identified. The different types of amyloidosis are named after the different protein precursor involved. It is important to recognize the type of amyloidosis because besides the general aspects of management, different types of amyloidosis have specific organ involvement, different pathophysiology, prognosis and treatment. The five most common protein precursors causing cardiac amyloidosis are light chain immunoglobulins (AL), heavy chain immunoglobulins, transthyretin amyloidosis (ATTR), serum amyloid A, and Apo lipoprotein A 1, and of these five, AL and ATTR are with much the most frequent proteins responsible for cardiac amyloidosis, mostly in the elderly.
The amyloid protein that most often infiltrates cardiac interstice is TTR and AL amyloid. Cardiac senile amyloidosis is related to deposits of TTR, and because of prognostic and therapeutic implications it is important to make differential diagnosis with other type of amyloidosis, particularly AL.6,9 Wild-type TTR is the more common cardiac amyloidosis, commonly known as cardiac senile amyloidosis, and less often is the mutant variety TTR that has its clinical expression earlier in life. TTR is synthesized mainly in the liver and less than 5% produced in the choroid plexus. It is a protein rich in beta stands responsible for physiological serum transportation of thyroxin and retinol, it occurs naturally in the form of a tetramer that does not misfold as monomer or adopt a beta pleated configuration except in the disease known as ATTR.12 Cardiac amyloidosis related to ATTR can occur because of: a) mutant ATTR of which at least 110 mutations have been described or, b) wild-type TTR11 (cardiac senile amyloidosis). TTR has an innate ability to aggregate into insoluble amyloid fibers and when it accumulates in tissues causing tissue and cell toxicity.13,14 With deposits of amyloid there is expansion of interstitial space, activation of inflammation and the presence of other amyloid attracted chaperon associated proteins present in all types of amyloidosis such as clustering, vimentin, vitronectin, pentraxin serum amyloid P (SAP), Apo lipoproteins, and glycosaminoglycans. Gradually there is loss of myocardial cells. There has been descriptions of different grades of inflammation, myocyte loss and extracellular expansion depending on the type of the amyloid infiltrating the heart. Because Amyloid heart disease is mainly an infiltrating interstitial heart disease, ventricles have increased thickness and are less compliant, there is progressive diastolic dysfunction that may be severe with very elevated right and left filling pressures, initially normal ejection fraction, but even at this early stages abnormal strain detected deformity, predominantly at basal zones sparing apical segments. There is a drop in cardiac output and associated autonomic neuropathy responsible for much of the symptoms in advanced stages of the disease.
Clinical presentation
Initially, amyloidosis is asymptomatic, as amyloid heart disease advances, there is progressive congestive symptoms and signs related initially to systemic and pulmonary venous hypertension related to the stiff, low compliant ventricles. In later stages, low cardiac output expresses itself as low blood pressure that with the associated autonomic neuropathy causes severe orthostatic hypotension and syncopal episodes. Ventricular and supraventricular arrhythmias and atrioventricular block are common. Typically, it is quite difficult to obtain that delicate balance between alleviating congestive signs and symptoms without precipitating data related to low cardiac output.
Carpal tunnel is a syndrome associated with ATTR. In 34% of patients with «idiopathic» carpal tunnel syndrome there is amyloid deposits in carpal tenosynovial tissues that typically antedates the cardiac diagnosis of ATTR by 8 to 10 years patients with a diagnosis of cardiac wild-type ATTR have carpal tunnel syndrome in 50% of the cases related to TTR deposit in the carpal tenosynovial tissues.14
Life expectancy is about 4 or 5 years in wild-type TR amyloidosis, and less than one year in light chain AL amyloid heart disease.
Diagnosis
As previously stated, cardiac amyloidosis is initially asymptomatic and if there is signs or symptoms of heart disease there is a tendency to relate it to other coexistent comorbidities of their advanced age. Somehow, there may be some clues that might suggest cardiac amyloidosis as an alternative or coexistent disease. Evidence of systemic infiltration of amyloid might suggest the presence of cardiac amyloid disease. Also as previously mentioned, if we see an elderly patient with HFPEF associated with carpal tunnel syndrome we should have in mind the possibility of amyloidosis.
Electrocardiograms (ECG) might disclose low voltage in standard and precordial leads in 36% of cases that is in discordance with the «hypertrophied» left ventricle disclosed in echocardiograms, there is an electro-echocardiographic discordance or mismatch that would be a clue for cardiac amyloidosis. In 65% of the patients there is Q waves in inferior leads or poor R-wave progression in anteroseptal leads simulating inferior or anteroseptal myocardial infarctions-«pseudo infarcts» (Figure 1). Also, there is commonly all type of supraventricular and ventricular arrhythmias including atrial fibrillation in 36% of patients.15 Ventricular tachycardia, different grades of atrioventricular block are also seen.
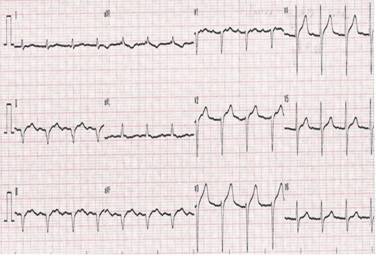
Figure 1: Electrocardiogram: inferior wall with «pseudo infarct» pattern, and poor R wave progression in right precordial leads.
X rays might show an enlarged cardiac silhouette with pericardial or pleural effusions.
Echocardiogram is the study more commonly used in patients with a suspected or confirmed diagnosis of cardiac amyloidosis. It gives us structural and functional information about eventual amyloid heart disease. Typically shows an increased thickness of left ventricular free wall and the septum that can be misdiagnosed as hypertrophic cardiomyopathy or hypertensive cardiomyopathy.16 In its classic echocardiographic presentation it has a speckled granular pattern that was the typical echocardiographic image, but know we know this pattern is quite unspecific and may appear in many other infiltrative heart disease, an also several patients with corroborated cardiac amyloidosis do not have this previously thought characteristic granular pattern, so even if it is a useful echocardiographic sign, we cannot rely on it for the diagnosis of cardiac amyloidosis. Other echo findings include increase thickness of atrioventricular valves, interatrial septum and free wall of right ventricle. Tissue Doppler e’ septal is low and E/e’ ratio is high, an indication of a high filling pressure of left ventricle (Figure 2). Left ventricular ejection fraction might be normal or minimally diminished, and a characteristic echo finding in cardiac amyloidosis is a diminished left ventricle longitudinal strain rate specifically in basal segments with preserved normal deformity in apical segments (Figures 3 and 4), there is a strain rate gradient from abnormal to normal from basal to apical segments.17,18
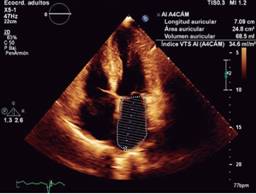
Figure 2: Echocardiogram: thick left ventricular wall with normal sized cavities. Enlarged atriums. Restrictive left ventricular filling as marker of increased filling pressure.
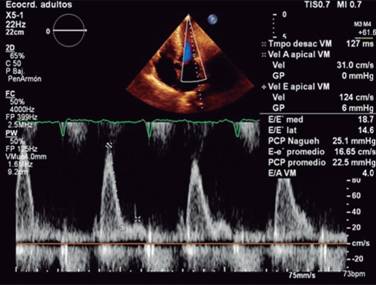
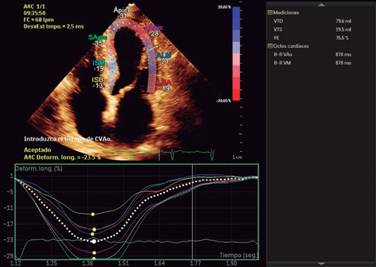
Figure 4: Strain imaging with depressed strain in basal segments of inferoseptal, inferior and inferolateral wall, with normal strain in apical segments, characteristic of cardiac amyloidosis.
Cardiac magnetic resonance (CMR) imaging gives important diagnostic and prognostic information regarding cardiac amyloidosis.19 CMR shows asymmetrical septal left ventricular hypertrophy (LVH) in 79% of patients with ATTR (70% sigmoid septum and 30% have reverse septal contour), symmetrical LVH occurs in 18% of patients and no LVH in 3%. Besides the gold standard structural and functional general information provided by a CMR, there is a characteristic late diffuse gadolinium enhancement in a subendocardiac or transmural pattern more or less typical of cardiac amyloidosis that may help differentiate it from other infiltrative or interstitial heart disease, in 29% there is subendocardial and 71% transmural gadolinium enhancement pattern.19,20 Preferably, in CMR it is better to use PSIR (phase sensitive inversion recovery) because technically it might be difficult to null myocardium and false negatives might result with other CMR acquisition sequence. Native T1 mapping (without gadolinium) and post contrast gadolinium help measure myocardial extracellular volume expansion associated with amyloidosis, information that gives additional prognostic information.21-26
An old test with new diagnostic indications is a pyrophosphate or biphosphonate based bone scan commonly used to detect metastatic bone disease. Cardiac amyloidosis has been recognized by radionuclide since 30 years ago.27 The usefulness of bone scintigraphy scans in differentiating the most common AL and TTR cardiac amyloidosis with the phosphate based radionuclide 99m-Tc-3,3-diphosphono-1,2-propanodicarboxylic acid (99m-Tc-DPD) appeared more than 10 years ago.28
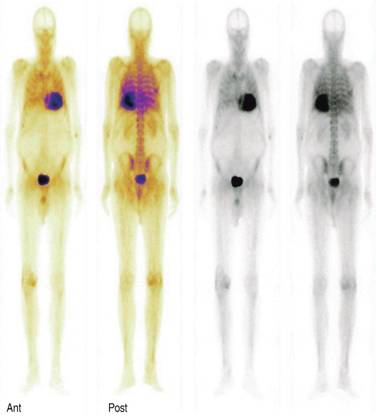
In bone scans we do not see cardiac uptake in normal hearts, that would be a Perugini uptake grade 0, when cardiac uptake is less than bone you say it is grade 1, uptake similar to bone is a grade 2, and when cardiac uptake is more than bone it is graded as 3 (Figure 5). Since several years ago it has been observed that in cardiac amyloidosis this type of bone scan discloses increased cardiac uptake of the radiotracer mainly in TTR amyloidosis, specifically the wild type TTR amyloidosis (WT-TTR). Several studies have clearly showed a sensitivity of more than 85% for WT-TTR and when we combine a positive cardiac uptake with a negative search for light chains in AL amyloidosis the sensitivity is practically a 100% for TTR amyloidosis. It is common practice to make a diagnosis of cardiac senile amyloidosis (WT-TTR) based exclusively on a positive grade 2 or 3 cardiac uptake of the bone radiotracer and a negative search for light chains in serum or urine. A false positive cardiac radiotracer uptake can be observed in AL cardiac amyloidosis, in such cases the cardiac bone scan radiotracer shows a mild grade 1 or 2 radiotracer uptake in one-third of patients with AL amyloidosis, however, these patients should have serological evidence of a monoclonal peak of light chain immunoglobulins.29 So with this information, it is possible to make the diagnosis of cardiac amyloidosis and specifically cardiac senile amyloidosis (WT-TTR) based on noninvasive test, obviating the need for a more invasive myocardial biopsy in this common frail population. Other important issue with phosphate based scans is the possibility to make early detection of cardiac involvement in ATTR, there is evidence of positive scans even before echocardiographic abnormalities are detected, an in addition 99m-Tc DPD has been compared with CMR for assessing cardiac amyloid infiltration and both methods are similar to identify myocardial amyloid infiltration, but the infiltration burden can be underestimated by visual analysis of CMR compared with 99m-Tc-DPD.30-35
Clinical outcome
In general, prognosis is not that favorable in cardiac amyloidosis. The clinical outcome depends in great part on the precursor amyloid protein precursor, being worse with AL amyloid that confers an average life expectancy of 9 months. TTR cardiac amyloidosis has a better prognosis with average survival of 3.5 to 5 years, in one study the median survival in cardiac ATTR after the onset of heart failure was 60 months, and in AL amyloid heart disease 5.4 months.36,37 Earlier diagnosis might have a favorable impact on prognosis, mainly with anti-plasma cell therapy in AL amyloid heart disease. There is still no clear cut treatment for TTR amyloidosis besides liver transplantation, but there are several ongoing trials that may impact long perm survival. It is assumed that preventing deposition of new amyloid fibrils in early diagnosed low amyloid loaded heart will impact prognosis better than trying to remove the usually insoluble protein fibrils in late diagnosed heavy loaded amyloid heart disease.
Management
Cardiac amyloidosis is a difficult to manage disease, and more when we see the patient in advanced very symptomatic stages. General measures usually include treatment of congestive heart failure with loop diuretics and aldosterone antagonists, but because these patients usually have thick walled small restrictive left ventricles we must be very cautious in titrating appropriate dose, even with mild diuresis they develop signs of low cardiac output with low blood pressure, orthostatic hypotension and syncope. Beta-blockers, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers are poorly tolerated and should be avoided.38 Digoxin binds to amyloid fibrils and can lead to locally high levels; it also must be used with caution.39-42
For patients with AL cardiac amyloidosis therapy is directed toward lowering kappa or lambda light chains mainly through lowering plasmatic cells mass with steroids, melphalan, proteasome inhibitors such as bortezomib, and other new developing therapies including stem cell transplant.
In ATTR amyloidosis there is no specific treatment, however, since the liver is responsible of synthesis of 95% of TTR, in hereditary mutant variety liver transplant have been done with success specially when cardiac TTR amyloidosis is detected in early stages preventing deposits of new deposits of the fibril amyloid TTR in the heart and slowing progression of the disease, this increases survival.
In wild-type ATTR general measures are used to control congestive symptoms or the more difficult to control low cardiac output or orthostatic hypotension, at this moment there is no clearcut modifying therapy for this complex disease. Modifying trials have been done that interfere or stabilize amyloid. There have been trials with interfering RNA to control the transcription of the abnormal DNA exon to diminish the production of TTR, also trials have been done with specific oligonucleotides directed against messenger RNA responsible for TTR translation, and results are not conclusive.
Stabilizing trials include epigallocatechin gallate, diflunisal, doxiciline plus taurodesoxicolic acid, tafamidis and others.
Epigallocatechin gallate, the most abundant catchin in green tea, inhibits amyloid fibril formation of several amyloid genic proteins. One observational study described 19 patients with ATTR cardiomyopathy who were serially evaluated with echocardiography and CMR after consuming green tea or green tea extracts, after 12 months no progression was detected.43
Diflunisal is a no steroidal anti-inflammatory drug (NSAID) that binds 99% of unoccupied thyroxin binding site in TTR, stabilizing familial TTR mutants against acid mediated fibrils monomers formation, it has been shown safe in patients with stable cardiac ATTR. The drawback is the known adverse effects NSAID has on renal sodium and water retention, hypertension, gastric effects, situations that might have deleterious results in an already hemodynamic compromised patients.44-48
Doxycycline acts inhibiting fibril formation and in combination with tauroursodeoxycholic acid might contribute to remove already deposited TTR protein. Preliminary phase II studies support a beneficial effect.49-51
TTR is normally a thyroxin transporter and has a specific binding site for this hormone. Tafamidis binds to the TTR binding sites for thyroxin, and it inhibits the degradation of the TTR tetramer into its monomer fibril-amyloid forming constituents. Tafamidis has showed slowing of polyneuropathy in early detected disease associated with ATTR and good safety issues after 5 year follow-up. Tafamidis has been approved in the European Union and in Japan for treatment of mutant ATTR. Analysis of the open-label extension of this study showed that long-term treatment with tafamidis was well tolerated with a reduced rate of neurological deterioration, effect sustained over 30 months. In a post-approval trial by the French Network for familiar amyloid polyneuropathy (FAP), neurological impairment scores worsened in 55% after 1 year of tafamidis treatment, suggesting that it could not stop disease progression (in terms of neural impairment) or disability in patients with advanced disease. Tafamidis is well tolerated, with few patients discontinuing treatment. Currently, tafamidis is not approved for use in the United States, there are undergoing trials actively recruiting patients. Tafamidis also prevents biochemical and echocardiographic decline in TTR cardiomyopathy.52-58
Conclusions
Cardiac senile amyloidosis is not infrequent as previously thought if we use the appropriate new noninvasive diagnostic tools well described with excellent sensitivity and specificity. Several of the current cases of HFPEF in the elderly might and are probably related to ATTR, a disease underdiagnosed and difficult to treat. This failure to find an appropriate treatment is in part related to misdiagnosis and also because small number of patients available for clinical trials that depend on histopathological diagnosis. Now with an old noninvasive study such as scintigraphy bone phosphate scan with this new diagnostic indication, much more patients with ATTR amyloidosis expressing itself as HFPEF will emerge and correctly diagnosed, this will bring interest in the disease and the direct consequence will be more therapeutic trials for this elderly related disease, and we have to remember that the elderly is a growing population that deserves our attention and should also be included in diagnostic and therapeutic trials.

