











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLas proteínas
En las células, el ácido desoxirribonucleico (DNA) almacena información hereditaria que permite transmitir características a la descendencia y contiene, entre otros datos, las secuencias codificadas de aminoácidos necesarias para producir proteínas (Rodríguez-Sotres, 2005). Por su parte, las proteínas actúan como las máquinas celulares encargadas de organizar y copiar al DNA y al ácido ribonucleico (RNA), degradar nutrientes para producir energía, sintetizar azúcares, lípidos, aminoácidos y casi todos los componentes de las células, transportan componentes de un sitio a otro, permiten el movimiento, detectan estímulos de luz, sonido y contacto con vecinos amigables u hostiles, y cumplen muchas otras funciones vitales (Figura 1). Dicho de otro modo, sin proteínas, no habría vida en la Tierra.
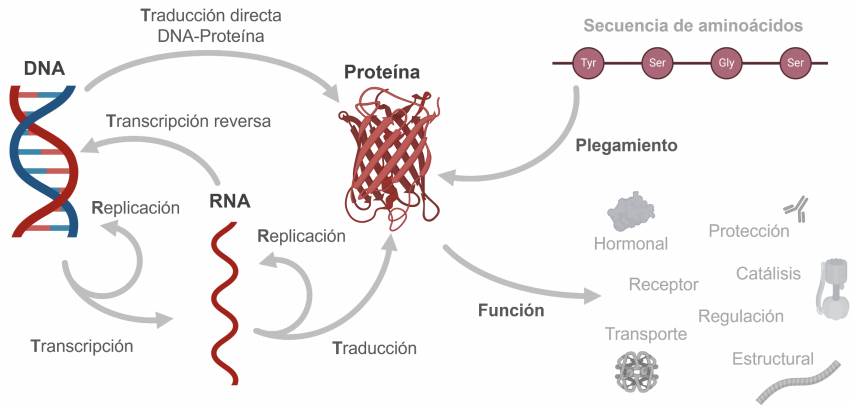
La información heredable se guarda en al DNA de las células. Mediante el proceso llamado transcripción fragmentos de información son copiados en RNA. La información en los RNA llamados mensajeros permite la traducción o síntesis de proteínas. Las proteínas son las dispositivos moleculares celulares encargados de casi todo lo demás.
Figura 1 El Dogma Central de la Biología Molecular.
Para poder hacer su trabajo, las proteínas deben adoptar una o más formas espaciales (arreglos tridimensionales) que llamamos plegamientos. Algunas proteínas se pliegan en una o pocas formas específicas, y se denominan estructuradas, mientras que otras adoptan múltiples formas, siendo estas últimas conocidas como intrínsecamente desestructuradas o desordenadas. La información para que una proteína se pliegue y adopte una forma espacial está en su secuencia de aminoácidos, según lo demostró Christian B. Anfinsen, quien recibió el premio Nobel de Química en 1972 (Anfinsen, 1973). Sin embargo, la relación es demasiado compleja debido a que los 20 diferentes aminoácidos pueden combinarse de muchas maneras, generando un número abrumador de secuencias posibles, muchas veces mayor al número de estrellas en el universo y que resulta imposible de explorar en su totalidad, al grado de que la naturaleza tampoco ha tenido oportunidad de hacerlo (Dill, 1999).
Al plegarse de una cierta manera, una proteína expone una superficie sinuosa que se complementa de manera específica con la de otras moléculas, grandes o pequeñas, que llamamos sus ligandos. Gracias a dicho reconocimiento entre moléculas la proteína encuentra su ubicación apropiada dentro de una célula y actúa de manera puntual sobre los ligandos que son de su competencia, sin afectar a los que no son reconocidos por esta. De este modo, aunque la célula contiene millones de substancias distintas, el hecho de que cada proteína interactúe solo con las moléculas adecuadas evita que la célula se desorganice, enferme o muera (Hartl y Hayer-Hartl, 2009), es decir: ¡Los plegamientos de una proteína son parte esencial de la función celular!
¿Cómo es que la secuencia de aminoácidos determina la forma?
Esa es justo la pregunta que nos encantaría contestar y que se conoce como el problema del plegamiento. Pero el problema es demasiado complejo y muchos detalles importantes parecen estar en el trayecto que la molécula sigue para plegarse, algo que conocemos como camino de plegamiento. Así, para conocer la forma de una proteína al detalle atómico se utilizan tres técnicas principales: Cristalografía de rayos X, Resonancia Magnética Nuclear y Criomicroscopía electrónica. Cada una con sus particularidades y limitaciones, pero tienen en común emplear equipos muy caros y requerir mucha cantidad de la proteína bien pura. En las tres técnicas, el trabajo es arduo, tardado y costoso. Además, las tres toman datos en tiempos largos, mientras que las proteínas se pliegan en fracciones de segundo. Esto significa que la forma que resolvemos es como una fotografía que da muy poca información del movimiento de la molécula. Aunque disponemos de numerosas “imágenes” tridimensionales de proteínas ya plegadas, sabemos poco sobre como alcanzaron esa forma (Mittag et al., 2010).
Así se explica la gran disparidad entre el número de proteínas cuya secuencia de aminoácidos se conoce y aquellas para las que también se conoce la estructura. En el caso de las proteínas del ser humano, en 2021, se contaba con información estructural para aproximadamente el 17% de ellas. Este conocimiento se podía extender hasta 40%, al considerar las proteínas de organismos estrechamente relacionados y con funciones equivalentes en nuestro organismo (Porta-Pardo et al., 2022).
Los Concursos Critical Assessment of protein Structure Prediction (CASP)
Dado que los métodos de determinación de estructuras son lentos, cuando se resuelve una nueva estructura, los datos se depositan en el portal de la base de datos Protein Data Bank (PDB) para ser revisados, mucho antes de ser publicados. En esa ventana de tiempo, los investigadores plantearon un desafío: “predecir la estructura de esas proteínas a partir de la secuencia”. Esto llevó a la creación del concurso internacional Critical Assessment of Structure Prediction (CASP), el cual busca evaluar a las estrategias de predicción de estructuras de proteínas e identificar a las más acertadas. El primer concurso CASP tuvo lugar en 1994 y, desde entonces, ha tenido lugar cada dos años. En cada edición, se han dado progresos graduales, pero los porcentajes de éxito en la predicción eran bajos y, hasta antes del CASP13 (luego de 28 años), estos rondaban el 31% (AlQuraishi, 2019).
A lo largo de esos concursos destacó el grupo de David Baker con una herramienta computacional llamada Rosetta. Este programa en realidad fue concebido para resolver el problema inverso, es decir: “si deseo obtener una proteína que tiene una cierta forma- ¿qué secuencia de aminoácidos le corresponde?” (Kuhlman et al., 2003). La herramienta fue mejorando y, con ella, David Baker ha sido capaz de producir una asombrosa cantidad de proteínas artificiales con formas novedosas que tienen aplicaciones biotecnológicas posibles (Figura 2) (DiMaio et al.,2011).
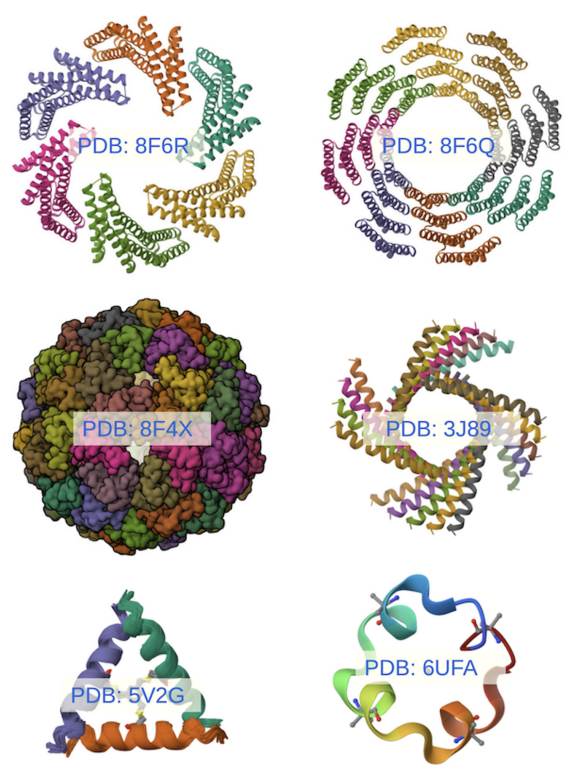
Para más detalles consulte los códigos indicados en cada imagen en el portal Protein Data Bank.
Figura 2 Esquema de la estructura de algunas proteínas artificiales diseñadas por el grupo de David Baker usando la herramienta Rosetta en sus diferentes versiones.
La herramienta de David Baker también tiene una aplicación sorprendente en el campo de la predicción de estructuras. Una proteína cuya estructura está en el PDB no requiere una predicción, ya conocemos su forma, pero ¿Cómo sabemos si cualquier predicción representa la estructura natural cuando no se ha resuelto su estructura?-Bueno, justo podemos usar la predicción y pedirle a Rosetta que nos diga cuáles secuencias de aminoácidos adoptarían esa forma-¡La secuencia de aminoácidos para la que predije esa forma debería aparecer en la lista de posibilidades que Rosetta nos da! Sí aparece, todo está bien, si no aparece, la predicción no es confiable. Esta estrategia se puede hacer cuantitativa con un poco de matemáticas y estadística (Martínez-Castilla y Rodríguez-Sotres, 2010).
Al CASP13 asistió un nuevo grupo financiado por una compañía denominada DeepMind™ y cofundada por Demis Hassabis en 2010, que fue adquirida por Google Inc. en 2014. Este grupo era un tanto ajeno al campo de las proteínas, pues su experiencia estaba en la programación de herramientas de Inteligencia Artificial (IA) para jugar videojuegos y juegos de mesa. Su éxito más sonado era el programa AlphaZero™ que pudo derrotar a su predecesor AlphaGo en un juego llamado Go y aprendió a jugar Ajedrez por sí mismo (Schrittwieser et al., 2020; Silver et al., 2018). Aunque solo compitieron en la categoría más difícil de las que incluye el desafío CASP, su herramienta AlphaFold superó a todos los equipos establecidos en esa categoría (AlQuraishi, 2019). Dos años más tarde, en el CASP14, este grupo incluyó la participación de John W. Jumper, quién se había incorporado a DeepMind y tenía bastante experiencia analizando información de proteínas en la computadora. En CASP14, AlphaFold 2.0 (AF2) rompió varios récords obteniendo 244 puntos, muy por encima de los 90.4 del segundo lugar. Su mejor predicción alcanzó una calidad de 92.8, la más alta registrada en la historia del concurso (Jumper et al., 2021a). El segundo lugar fue justamente el grupo de David Baker con su herramienta mejorada RoseTTAFold, la cual ya añadía herramientas de IA a su estrategia original (Baek et al., 2021).
La herramienta AlphaFold 2.0
AF2 es una herramienta basada en IA, entrenada con dos piezas de información fundamentales. Por un lado, se entrenó con las estructuras conocidas de las proteínas que han sido depositadas en la base de datos PDB. Por otro lado, se capacitó para comparar la secuencia de aminoácidos cuyo plegamiento se desea predecir con todas las secuencias de proteínas similares y que deben tener funciones parecidas, para luego extraer la información que llamamos evolutiva. Es decir, el programa analiza cómo las proteínas van cambiando a lo largo de la evolución de las especies y encuentra los sitios en que no hay cambios (sitios conservados), aquellos que cambian mucho (sitios variables) y aquellos en los que un cambio puede ocurrir, siempre que sea compensado por un cambio en otro sitio de la misma secuencia (sitios conectados). De esa manera, la Red Neural Profunda (la inteligencia dentro del programa) determina cuáles aminoácidos deben estar cerca uno de otro al doblarse la proteína para tomar su forma funcional (llamada plegamiento nativo). A partir de esa información, empaqueta los aminoácidos en el espacio siguiendo las reglas que aprendió de las estructuras conocidas al “estudiar” el PDB (Jumper et al., 2021b). Suena complicado y lo es, debido a la gran cantidad de información y a los muchos detalles involucrados. Sin embargo, las computadoras son capaces de procesar grandes volúmenes de datos de manera rápida y precisa.
Beneficios para la humanidad
Tras su inusitado éxito en el concurso CASP14, DeepMind se dio a la tarea de utilizar esta herramienta para predecir la estructura del proteoma humano completo, lo que abarca todas las proteínas identificadas en el genoma humano (Tunyasuvunakool et al., 2021). Posteriormente, predijeron todas las estructuras en la base de datos de UniRef100, que contiene cientos de millones de secuencias de aminoácidos derivados de los genomas de centenas de organismos diferentes cuyos genomas han sido secuenciados (Varadi et al., 2022; Varadi et al., 2023). Desde luego, aquí se incluyen animales como el humano, el ratón, el chimpancé, el cerdo, el elefante, por citar algunos; plantas como el maíz, sorgo, caña de azúcar, trigo, cebada, tomate, papa y muchas más; una gran variedad de hongos, así como muchísimas bacterias y un gran número de virus. Como ejemplo, la Figura 3 muestra el portal correspondiente a la predicción para la proteína humana enzima convertidora de angiotensina 2 (ACE2), que es la puerta de entrada para el virus causante de la enfermedad de COVID19 (SARS-CoV2). La contribución desinteresada a la humanidad es de destacarse, puesto que el financiamiento viene de una compañía comercial. Por otro lado, AF2 y RoseTTAFold son también programas liberados para el uso académico y de investigación (Mirdita et al., 2022).
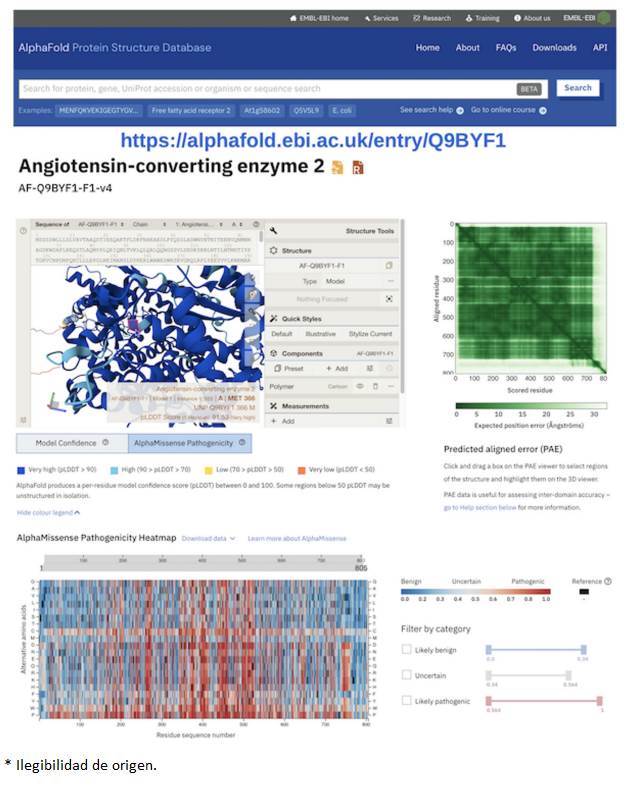
En la parte inferior se muestra la información generada por otra IA relacionada llamada AlphaMisssense que clasifica las mutaciones del gen que podrían causar enfermedad en el ser humano al afectar el plegamiento y/o la función de la proteína.
Figura 3 Predicción por AlphaFold de la estructura de la enzima convertidora de Angiotensina 2 humana (la puerta de entrada del SARS-CoV-2, causante del COVID-19) disponible en el recurso web alphafold.ebi.ac.uk con el código A0A7I2V5W5.
En México y América Latina, varios grupos de investigación han venido empleado estas herramientas desde sus inicios y en esta área la UNAM ha tenido una participación destacada. Los estudios incluyen proteínas artificiales (Romero-Romero et al., 2021; León-González et al., 2022), proteínas relacionadas con padecimientos neurodegenerativos (Guzman-Ocampo et al., 2023), diseño de nuevos fármacos (Padilla-Mayne et al., 2024), rediseño de enzimas para biorremediación (Aguirre-Sampieri et al., 2024) y creación de biosensores proteicos (González-Andrade et al., 2013), por mencionar algunos.
La investigación se está moviendo rápidamente para incorporar nuevas potencialidades a la herramienta de predicción de estructura de proteínas (Ozden et al., 2023). DeepMind puso también a disposición de la comunidad académica una herramienta más desarrollada, capaz de predecir ya no solo la forma de una proteína aislada, sino la manera en que se asocia con sus ligandos, nombrada AlphaFold3 (Abramson et al., 2024). Esta herramienta promete ser muy poderosa, pero la comunidad aún está aprendiendo a explotarla y no conocemos suficientemente sus limitaciones. En este caso, es posible que la herramienta no sea completamente gratuita, ya que al predecir la interacción de proteínas con moléculas pequeñas se puede aplicar al diseño de fármacos y las grandes empresas farmacéuticas estarán dispuestas a pagar millones de dólares para conocer la función de fármacos de modo confiable y preciso. Esperemos que los académicos podamos usarla por un precio más modesto. Por su parte, el grupo de David Baker ha anunciado la versión All-Atom de su herramienta RoseTTaFold (Krishna et al., 2024), la cual también puede predecir la interacción de proteínas con moléculas pequeñas, permitiendo diseñar proteínas artificiales para reconocer moléculas específicas. Sin duda una herramienta con gran poder en desarrollos de nuevas aplicaciones biomédicas biotecnológicas.
Conclusión
El importante y amplio impacto de las contribuciones de Demis Hassabis, John M. Jumper y David Baker al estudio de la sorprendentes y sofisticadas máquinas moleculares de la vida que llamamos proteínas justifica, sin duda, el otorgamiento del Premio Nobel de Química a estos tres investigadores.
Vale la pena destacar que no existe una receta para ser galardonado con el premio Nobel, David Baker cuenta con más de 30 años de liderazgo científico de muy alto nivel, más de 500 publicaciones y un índice h de 146, que refleja el impacto significativo de su extenso trabajo en la comunidad académica y científica. En cambio, John M. Jumper tiene menos de 3 años como investigador independiente, 19 publicaciones y un índice h de 9, pero si bien reciente, su contribución ha sido muy significativa.
Una importante lección para los jóvenes es que no existe una manera de forzar a que la ciencia avance en la dirección que se nos antoje, los avances se logran paso a paso y los hitos más significativos suceden en áreas muy diversas. Todavía no hay una herramienta de IA ni de otro tipo capaz de predecir cuál será el siguiente gran descubrimiento de la humanidad.

