











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El género Vanilla comprende alrededor de 130 especies distribuidas en las zonas tropicales del mundo, en su mayoría son originarias del continente americano (Azofeifa-Bolaños et al., 2017). En México se reportan 10 especies del género Vanilla; sin embargo, algunos trabajos recientes de caracterización morfológica y genética indican que la diversidad genética de vainilla en México puede
En plantaciones comerciales de vainilla la variabilidad genética se considera limitada (Hernández-Leal et al., 2016), principalmente por la reproducción vegetativa utilizada para establecer nuevos huertos, la polinización manual que se realiza para la obtención de frutos y la limitada germinación de las semillas; además, la falta de nuevos genotipos provenientes de la recombinación sexual ha acentuado aún más la erosión genética en las plantaciones de vainilla (Schlüter et al., 2007); sin embargo, se ha observado en huertos de vainilla una variación morfológica, sobre todo en las hojas, aun en el mismo individuo, características distintivas presentes en las plantas hemiepífitas y algunas monocotiledóneas (Ray, 1990), variabilidad que puede ser usada como fuente de germoplasma en programas de mejoramiento genético de este cultivo (Soto y Dressler, 2010).
La diversidad genética se refiere a las variaciones heredables que ocurren en cada organismo, entre los individuos de una población y entre las poblaciones dentro de una especie en condiciones naturales más o menos estables (Rimieri, 2017). En este sentido, diversos trabajos se han realizado para conocer la diversidad genética del género Vanilla en diferentes niveles y con diferentes objetivos. Cameron (2018) y Ramos-Castellá et al. (2017) realizaron estudios genotípicos, fenotípicos y bioquímicos para obtener información sobre su evolución y características biológicas; Ferreira et al. (2020), Flanagan et al. (2018) y Parizaca (2019) han propuesto mediante caracterización genética y fenotípica nuevas especies del género Vanilla, mientras que Martínez-Castillo et al. (2007) han realizado trabajos sobre relaciones filogenéticas y Nagy et al. (2012) sobre determinaciones taxonómicas.
Los marcadores moleculares que hacen uso de la reacción en cadena de la polimerasa (PCR), como las inter secuencias simples repetidas, o ISSR (Zietkiewicz et al., 1994) presentan algunas ventajas al compararlos con los descriptores morfológicos y bioquímicos. Los ISSR discriminan entre individuos de especies genéticamente cercanas o entre variedades de la misma especie (Reyes-Alemán et al., 2013); además, los ISSR generan información valiosa en especies que no han sido estudiadas desde el punto de vista genético y son una técnica simple y económica (Araújo et al., 2016).
En un estudio realizado para determinar las relaciones genéticas de siete especies y dos híbridos de vainilla se utilizaron 10 marcadores tipo ISSR y determinaron que hubo una relación cercana entre individuos de V. planifolia, V. tahitensis y V. aphylla, asociada con bajos niveles de variación (Verma et al., 2009); así mismo, determinaron la relación filogenética entre V. planifolia y otras tres especies de vainilla en México, corroborando que hay diferencias claras entre V. planifolia, V. pompona, V. insignis y V. odorata. En tanto, Ramos-Castella et al. (2017) en un estudio relacionado utilizaron cinco cebadores ISSR.
Villanueva-Viramontes et al. (2017) utilizaron ISSRs para discriminar entre individuos silvestres de V. planifolia y otras especies que crecen naturalmente en la península mexicana de Yucatán (PMY). Estos investigadores encontraron que los ISSR pueden identificar especies de vainilla presentes en la PMY debido a los altos niveles de variación genética en las vainillas estudiadas.
El material biológico utilizado en las plantaciones comerciales de vainilla en México se basa en clones nativos de V. planifolia, ya que aún no se han generado variedades mejoradas en esta especie. Conocer la diversidad genética intra e interespecífica en cultivos útiles para el hombre es de gran importancia para consolidar un programa de mejoramiento genético en sus primeras etapas, como una estrategia para ayudar a resolver problemas de tipo técnico, biológico y climático, es por esto que el objetivo del presente estudio fue estimar la diversidad genética en híbridos intraespecíficos de cruza simple de V. planifolia, así como en accesiones parentales con datos moleculares obtenidos de cuatro marcadores tipo ISSR.
Materiales y métodos
Material vegetal
El material biológico utilizado estuvo compuesto por 38 genotipos de Vanilla planifolia del banco de germoplasma de vainilla de la Benemérita Universidad Autónoma de Puebla (BUAP) (Cuadro 1), ubicado en el municipio de Tenampulco, Puebla, México, en las coordenadas geográficas 20° 11’ 52.23’’ Lat. N y 97° 22’ 06.09’’ Long. O y altitud de 224 msnm. Se colectaron hojas de plantas de ocho años de edad de 16 accesiones provenientes de los estados de Chiapas, Puebla, Quinta Roo y Veracruz, México, cada procedencia constituyó una población; 21 híbridos intraespecíficos de cruza simple de dos años de edad conformaron la quinta población, el testigo fue un clon de V. planifolia comúnmente cultivado en huertos comerciales de vainilla.
Cuadro 1 Material biológico utilizado para el estudio de diversidad genética en híbridos de cruza simple y accesiones parentales de V. planifolia de México.
| Accesión | Origen | Cruza | Cruza |
| 2 | Ejido La Corona, Márquez de Comillas, Chiapas | × 41 | × 21 |
| 16 | San Javier, Ocosingo, Chiapas | × 124 | × 20 |
| 20 | Nuevo Becar, Othón P. Blanco, Quintana Roo | × 39 | × 28 |
| 21 | Nuevo Becar, Othón P. Blanco, Quintana Roo | × 2 | × 35 |
| 28 | Tenampulco El Viejo, Tenampulco, Puebla | × 22 | × 111 |
| 35 | La Palapa, Tenampulco, Puebla | × 69 | Testigo |
| 36 | Primero de Mayo, Papantla, Veracruz | × 112 | |
| 39 | Primero de Mayo, Papantla, Veracruz | × 2 | |
| 41 | Segundo Cantón Fracción Villanueva, Tapachula, Chiapas | × 194 | |
| 69 | Huatusco, Othón P. Blanco, Quintana Roo | × 2 | |
| 71 | Huatusco, Othón P. Blanco, Quintana Roo | × 39 | |
| 89 | Caracoles, Tenampulco, Puebla | × 21 | |
| 111 | Miahuatlán, Tihuatlán, Veracruz | × 21 | |
| 112 | Copales Arroyo Blanco, San José Acateno, Puebla | × 16 | |
| 115 | San Rafael, Veracruz | × 27 | |
| 124 | Tihuatlán, Veracruz | × 28 |
Para la extracción de ADN se utilizó tejido vegetal (hojas sanas) de cada genotipo, el cual fue colectado en el material de campo del banco de germoplasma. Se desinfectó con alcohol etílico 96 % para eliminar polvo e impurezas, las muestras se colocaron en bolsas de papel y éstas se colocaron en hielo para su conservación y trasladadas a la BUAP. Posteriormente, las muestras fueron transportadas al Laboratorio de Biotecnología de Plantas, Escuela de Ciencias Biológicas de la Universidad Nacional de Costa Rica para los análisis moleculares.
Extracción de ADN
El protocolo de extracción empleado fue una modificación del método basado en el detergente catiónico bromuro de cetiltrimetilamonio (CTAB), incluyendo un tratamiento con ARNasa, seguido de una extracción con cloroformo y una precipitación con isopropanol (Rasoamanalina et al., 2023; Stewart, 1997,). En un tubo de microcentrífuga de 1.5 mL se introdujo un segmento de hoja de 50-60 mg, se agregaron dos balines metálicos con diámetro de 3 mm y 400 µL de solución de homogenización (100 mM Tris-HCl, pH 8.0, 20 mM EDTA, 2 % p/v CTAB, 1.42 M NaCl, 2.4 μL β-mercaptoetanol, 0.1 mg mL-1 proteinasa K). Los tubos se colocaron en el disruptor de tejidos (MM400 (Retsch GmbH, Haan, Alemania) por 3 min a 30 Hz para macerar la muestra hasta obtener una mezcla homogénea. Las muestras fueron incubadas a 60 ºC durante 60 min, agitando los tubos por inversión cada 10 min. Se agregaron 3 µL de RNasa (20 mg mL-1) y se incubó por 15 min a 37 ºC. Posteriormente, se agregaron 500 µL de una mezcla de cloroformo:octanol (24:1 v/v) y se homogeneizó por inversión del tubo durante 1 min; las muestras se centrifugaron a 15000 rpm durante 10 min, el sobrenadante se transfirió a un nuevo tubo. El ADN se precipitó con 1 v de isopropanol, se mezcló por inversión y se volvió a centrifugar a 15000 rpm por 10 min. El precipitado de ADN se lavó con 500 µL de etanol 70 % y se centrifugó nuevamente. Se removió el etanol con micropipeta y el precipitado de ADN se dejó secar a temperatura ambiente con el tubo invertido por 15 min. Finalmente, el ADN se resuspendió en 50 µL de solución Tris 10 mM (pH 8.0), las trazas de etanol fueron evaporadas incubando los tubos a 45 ºC durante 3 min; las muestras fueron almacenadas a -20 ºC hasta su uso.
La calidad del ADN se determinó por electroforesis horizontal en geles de agarosa 1 %. La electroforesis se realizó en una solución amortiguadora de corrido SB 1X (sodio-borato) con una fuente de poder ENDURO E0303 (Labnet, Edison, Nueva Jersey, EUA) a 120 V durante 40 min. Como marcador de peso molecular se utilizó GeneRuler 100 bp Plus DNA Ladder (Thermo Scientific, Carlsbad, California, EUA). El gel se observó en un transiluminador con luz ultravioleta modelo ENDURO GDS (Labnet, Edison, Nueva Jersey, EUA). La concentración y pureza del ADN se estimaron en un espectrofotómetro (NanoDrop 2000, Thermo Scientific, Carlsbad, California, EUA) y se utilizaron las relaciones de absorbancia 260/280 y 260/230 como indicadores de la pureza.
Marcadores ISSR, amplificación de PCR y electroforesis
Para evaluar la diversidad de híbridos y accesiones parentales se evaluaron 10 marcadores ISSR (Verma et al., 2009), de los cuales, cuatro presentaron alto nivel de polimorfismo. Se trabajaron reacciones de PCR con un volumen final de 20 µL con las siguientes concentraciones finales: 30 ng de ADN, solución amortiguadora 1X [10 mM Tris-HCl (pH 8.8 a 25 °C), 50 mM KCl, 0.08 % (v/v) Nonidet P40], 0.2 mM de dNTPs, 2 mM de MgCl2, 0.5 mM de cada iniciador, 1 U de Taq polimerasa y agua grado molecular.
La amplificación por PCR se llevó a cabo en un termociclador (modelo ProFlex PCR System, Applied Biosystems, Singapur) usando el siguiente programa: desnaturalización inicial a 94 °C por 5 min, seguido por 40 ciclos de 40 s a 94 °C para la desnaturalización, 45 s a la temperatura de hibridación específica para cada iniciador y 90 s a 72 °C para la extensión. La temperatura de hibridación fue: 50 ºC para los iniciadores ISSR-C07 (5’-GAGAGAGAGAGAGAGAC-3’) e ISSR-T06 (5’-AGAGAGAGAGAGAGAGT-3’), 57 y 61 ºC para ISSR-C09 (5’-CAGATGGGAGTCAAGTCAAC-3’) e ISSR-C10 (5’-ACCTCCTGCAGATTCGTGTC-3’), respectivamente. La reacción concluyó con una extensión final a 72 °C por 10 min. Posteriormente, los fragmentos amplificados fueron separados electroforéticamente en gel de agarosa 2 % a 100 V durante 2 h. La post-tinción del gel se realizó con Gel Red (Biotium, San Francisco, California, EUA), siguiendo las recomendaciones del fabricante. Finalmente, la observación de fragmentos se llevó a cabo en un transiluminador ENDURO GDS (Labnet, Edison, Nueva Jersey, EUA).
Análisis estadístico
A partir de la información genotípica se generó una matriz binaria donde 1 indicaba presencia y 0 ausencia del fragmento, para medir la capacidad informativa de los marcadores ISSR y su utilidad. Para evaluar la diversidad genética, se estimaron tres parámetros: contenido de información polimórfica (CIP) (Roldan-Ruiz et al., 2000), índice de marcador (IM) (Varshney et al., 2007) y poder de resolución (PR) (Prevost y Wilkinson, 1999). El valor CIP de cada locus se estimó usando la fórmula:
Donde f i es la frecuencia del locus o banda i en la población; por lo tanto, para conocer el CIP de cada marcador ISSR se calculó el promedio de todos los loci de cada marcador.
La relación múltiple efectiva (RME) se estimó usando la fórmula: (Varshney et al., 2007)
Donde n es el número promedio de fragmentos amplificados por cada accesión por marcador y β la proporción de loci polimórficos (LP) respecto al total de loci obtenidos por cada marcador, la cual se obtuvo con la relación LP/número total de loci. El IM se calculó como:
El PR de cada marcador se estimó de acuerdo con Prevost y Wilkinson (1999), usando la fórmula:
Donde Ib = 1 - (2 × | 0.5 - pi |), y pi es la proporción de accesiones que contienen la i-ésima banda.
El análisis de varianza molecular (AMOVA) se realizó en el programa GenAlEx 6.5 (Peakall y Smouse, 2012). Dicho análisis se realizó primero considerando solamente las accesiones parentales y posteriormente progenitores e híbridos. El análisis de componentes principales se llevó a cabo con la matriz de datos binarios y usando el paquete factoextra (Lemenkova, 2019), en el programa R versión 3.5.2. El agrupamiento de los genotipos se evaluó por medio de las pruebas Gap (Tibshirani et al., 2009) y Elbow (Thorndike, 1953) utilizando el paquete factoextra. Las relaciones de similitud entre los genotipos se establecieron a través del análisis de conglomerados con datos estandarizados. Las distancias genéticas de Jaccard y el criterio de medias aritméticas no ponderadas (UPGMA) se usaron para generar un dendrograma en el programa PAST versión 2.17 (Hammer et al., 2001). La asociación de cada agrupamiento se evaluó por el método de remuestreo multiescalar bootstrap con 10,000 repeticiones.
Para estudiar la estructura genética de vainilla se realizó un análisis de agrupamiento bayesiano en el programa Structure 2.3.4 (Pritchard et al., 2000). Previo al análisis, se fijaron los parámetros de modelo mezclado y frecuencia de alelos correlacionados. El número óptimo de agrupaciones se evaluó de 1 a 10, con 10 simulaciones independientes por cada valor de K, 100,000 cadenas de Markov Monte Carlo y 10,000 cadenas iniciales excluidas. El número de agrupaciones poblacionales se determinó con el método de Evanno et al. (2005) usando el programa Structure Harvester v. 0.6.93 (Earl y vonHoldt, 2012).
Resultados y discusión
Polimorfismo y capacidad resolutiva de marcadores ISSR en vainilla
El marcador ISSR-C10 generó el mayor número de bandas polimórficas, con un total de 42; por otra parte, ISSR-T06 y ISSR-C07 fueron los que generaron el número de bandas más bajo, 18 y 25, respectivamente. En estudios previos donde se usaron marcadores RAPD y AFLP en vainilla se ha reportado el uso eficiente de tales marcadores para el estudio de la diversidad genética (Besse et al., 2004; Divakaran et al., 2006). En este estudio se observó que ISSR-C10 y ISSR-C09 tuvieron el poder resolutivo más alto (Cuadro 2), lo cual indica que ambos marcadores son eficientes para el estudio de la diversidad de vainilla, ya que pueden detectar diferencias entre una cantidad grande de genotipos (Prevost y Wilkinson, 1999).
Cuadro 2 Capacidad de cuatro marcadores ISSR para estimar la diversidad genética de V. planifolia.
| Iniciadores | Secuencia | NB | BP | BNP | PR | CIP | IM |
| ISSR-C07 | 5’GAGAGAGAGA GAGAGAC3’ | 25 | 24 | 1 | 9.87 | 0.28 | 1.29 |
| ISSR-T06 | 5’AGAGAGAGA GAGAGAGT3’ | 18 | 18 | 0 | 9.08 | 0.31 | 1.51 |
| ISSR-C09 | 5’CAGATGGGA GTCAAGTCAAC3’ | 33 | 33 | 0 | 12.92 | 0.29 | 1.84 |
| ISSR-C10 | 5’ACCTCCTGCA GATTCGTGTC3’ | 42 | 42 | 0 | 17.08 | 0.30 | 2.55 |
NB: número de bandas generadas, BP: bandas polimórficas, BNP: bandas no polimórficas, PR: poder de resolución, CIP: contenido de Información polimórfica, IM: índice de marcador.
El contenido de información polimórfica (CIP) mide la capacidad discriminatoria de un marcador, la cual depende del número de alelos conocidos (establecidos) y su frecuencia de distribución (Botstein et al., 1980). En este estudio, el ISSR-T06 fue el marcador con el valor más alto (0.31); por el contrario, el ISSR-C07 generó menos información polimórfica (CIP = 0.28) (Cuadro 2). Estos valores fueron similares a los obtenidos por Verma et al. (2009), quienes reportaron valores de CIP cercanos a 0.30 para los mismos marcadores ISSR.
Por otro lado, gran parte de la población estuvo compuesta por genotipos clasificados como V. planifolia, por lo que el marcador ISSR-T06 detectó un alto polimorfismo entre individuos de la misma especie. En este sentido, Verma et al. (2009) reportaron que el ISSR-T06 fue el más polimórfico entre ocho especies de vainilla, lo que sugiere que el ISSR-T06 puede proveer información importante de similitud genética entre y dentro de especies de vainilla.
El índice de marcador corresponde al producto del CPI y el número de loci polimórficos, y puede ser utilizado como una aproximación para determinar la utilidad de un marcador (Varshney et al., 2007). En este estudio se observó que ISSR-C10 y ISSR-C09 presentaron el mayor índice de marcador (2.55 y 1.84, respectivamente), lo que refuerza su aplicación en el estudio de diversidad de la vainilla.
Diversidad genética en vainilla
El AMOVA reveló alta varianza genética (97 %) dentro de las poblaciones y poca varianza (3 %) entre las poblaciones (Cuadro 3) considerando genotipos parentales de diferente origen geográfico e híbridos de cruza simple, los cuales constituyeron las poblaciones. Estos resultados indicaron que existe una mayor diferenciación genética dentro de poblaciones de vainilla que entre poblaciones, debido a que las accesiones parentales fueron de diferente origen geográfico y los híbridos crean nuevas combinaciones genéticas al realizar las cruzas. Un resultado similar fue reportado por Schlüter et al. (2007), quienes encontraron mayor variación genética en V. planifolia dentro de poblaciones que entre poblaciones. Conocer la diversidad genética entre y dentro de poblaciones es importante para el diseño de un programa de mejoramiento genético por selección e hibridación.
Cuadro 3 Análisis molecular de varianza en la subpoblación parental y población total de V. planifolia de México.
| AMOVA considerando genotipos parentales e híbridos | |||||
| Fuente | GL | SC | CM | Estimación de varianza | % de varianza |
| Entre poblaciones | 4 | 87.24 | 21.81 | 0.59 | 3 |
| Dentro de poblaciones | 33 | 610.18 | 18.49 | 18.49 | 97 |
| Total | 37 | 697.42 | 19.08 | 100 | |
| AMOVA considerando genotipos parentales | |||||
| Entre poblaciones | 3 | 49.08 | 16.36 | 0.00 | 0 |
| Dentro de poblaciones | 11 | 189.05 | 17.19 | 17.19 | 100 |
| Total | 14 | 238.13 | 17.19 | 100 | |
Se obtuvo un índice poblacional promedio informativo de Shannon de 0.28 (Cuadro 4). Considerando sólo las accesiones parentales, en la mayoría de las variables de los índices de diversidad se observó que las poblaciones de Puebla y Veracruz, México registraron valores altos, debido a que la región del Totonacapan (Puebla y Veracruz) es considerada como la zona donde se comenzó a cultivar V. planifolia (Bory et al., 2008; Lubinsky et al., 2008) y en la actualidad son los principales estados productores de México (Luis-Rojas et al., 2020). Estos resultados indican que el material biológico de Puebla y Veracruz tiene potencial para iniciar un programa de mejoramiento genético por selección. Por otro lado, el grupo de híbridos superó los índices de diversidad genética de las poblaciones parentales, lo que sugiere que la hibridación es una estrategia que genera buenos resultados en la creación de material biológico con nuevas combinaciones genéticas con características agronómicas deseables. Los valores del índice de Shannon variaron de 0.21 a 0.30, menores que los reportados por Verma et al. (2009), quienes obtuvieron valores superiores a 0.5 con RFLPs e ISSR.
Cuadro 4 Índices de diversidad de las cinco poblaciones de V. planifolia , fijadas de acuerdo con el origen geográfico y origen genético.
| Población | % LP | NAD | EE | NAE | EE | IIS | EE | HE | EE | HEI | EE |
| Chiapas | 38.26 | 0.78 | ± 0.09 | 1.23 | ± 0.03 | 0.21 | ± 0.03 | 0.14 | ± 0.02 | 0.17 | ± 0.02 |
| Quintana Roo | 56.52 | 1.14 | ± 0.09 | 1.25 | ± 0.03 | 0.26 | ± 0.02 | 0.16 | ± 0.02 | 0.19 | ± 0.02 |
| Puebla | 55.65 | 1.15 | ± 0.09 | 1.32 | ± 0.03 | 0.30 | ± 0.03 | 0.20 | ± 0.02 | 0.24 | ± 0.02 |
| Veracruz | 55.65 | 1.12 | ± 0.09 | 1.25 | ± 0.03 | 0.26 | ± 0.02 | 0.17 | ± 0.02 | 0.18 | ± 0.02 |
| Híbridos | 92.17 | 1.84 | ± 0.05 | 1.33 | ± 0.02 | 0.36 | ± 0.02 | 0.22 | ± 0.01 | 0.23 | ± 0.01 |
% LP: porcentaje de loci polimórficos, NAD: número de alelos diferentes, EE: error estándar, NAE: número de alelos efectivos, IIS: índice informativo de Shannon, HE: heterocigocidad esperada, HEI: heterocigocidad esperada insesgada.
En el análisis de componentes principales se observó que los dos primeros componentes explicaron el 15.7 % de la diversidad, lo cual se considera bajo, ya que fueron necesarios al menos 10 componentes para poder explicar más de 50 % de la diversidad genética presente en la población de vainilla estudiada (Figura 1). No obstante, de acuerdo con las pruebas Gap (Tibshirani et al., 2009) y Elbow (Thorndike, 1953), se observaron dos grupos dentro de la población. El primer grupo estuvo conformado primordialmente por genotipos parentales, mientras que el segundo grupo estuvo integrado por los híbridos. En el primer grupo los genotipos parentales no formaron subgrupos con base en su origen geográfico, lo cual indicó que los genotipos de Veracruz, Puebla, Quintana Roo y Chiapas, México presentan alta similitud genética, debido a que la reproducción comercial para el establecimiento de nuevos huertos de vainilla se realiza por esquejes, y generalmente los productores de Quintana Roo y Chiapas adquieren material biológico de Veracruz y Puebla. Estas interrelaciones genéticas entre poblaciones de V. planifolia originarias de los estados de Veracruz y Quintana Roo también han sido reportadas por Ramos-Castellá et al. (2017).
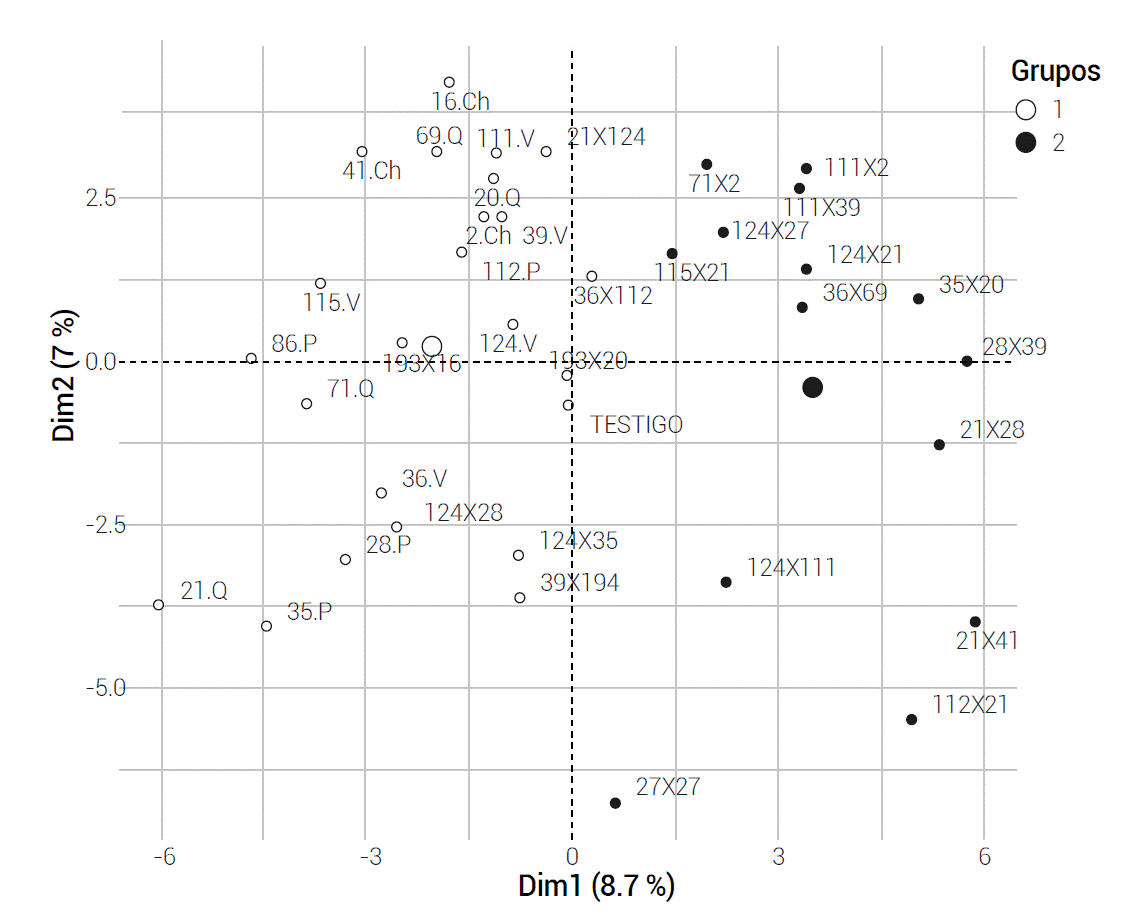
Figura 1 Dispersión de los 38 genotipos de V. planifolia agrupados de acuerdo con los dos primeros componentes principales (Dim). Las agrupaciones se realizaron en función de las pruebas de Gap (Tibshirani et al., 2009) y Elbow(Thorndike, 1953). Q: Quintana Roo, V: Veracruz, P: Puebla, Ch: Chiapas.
De acuerdo con las distancias genéticas de Jaccard, los genotipos con mayor similitud fueron las cruzas 111 × 2 y 111 × 39, con un valor de 0.54, mientras que el más alejado genéticamente fue el progenitor 111. En el análisis de agrupamiento, la prueba de remuestreo bootstrap detectó bajos niveles de asociación entre los genotipos para la formación de grupos (Figura 2), estos resultados indican la presencia de una alta similitud genética entre los genotipos evaluados.
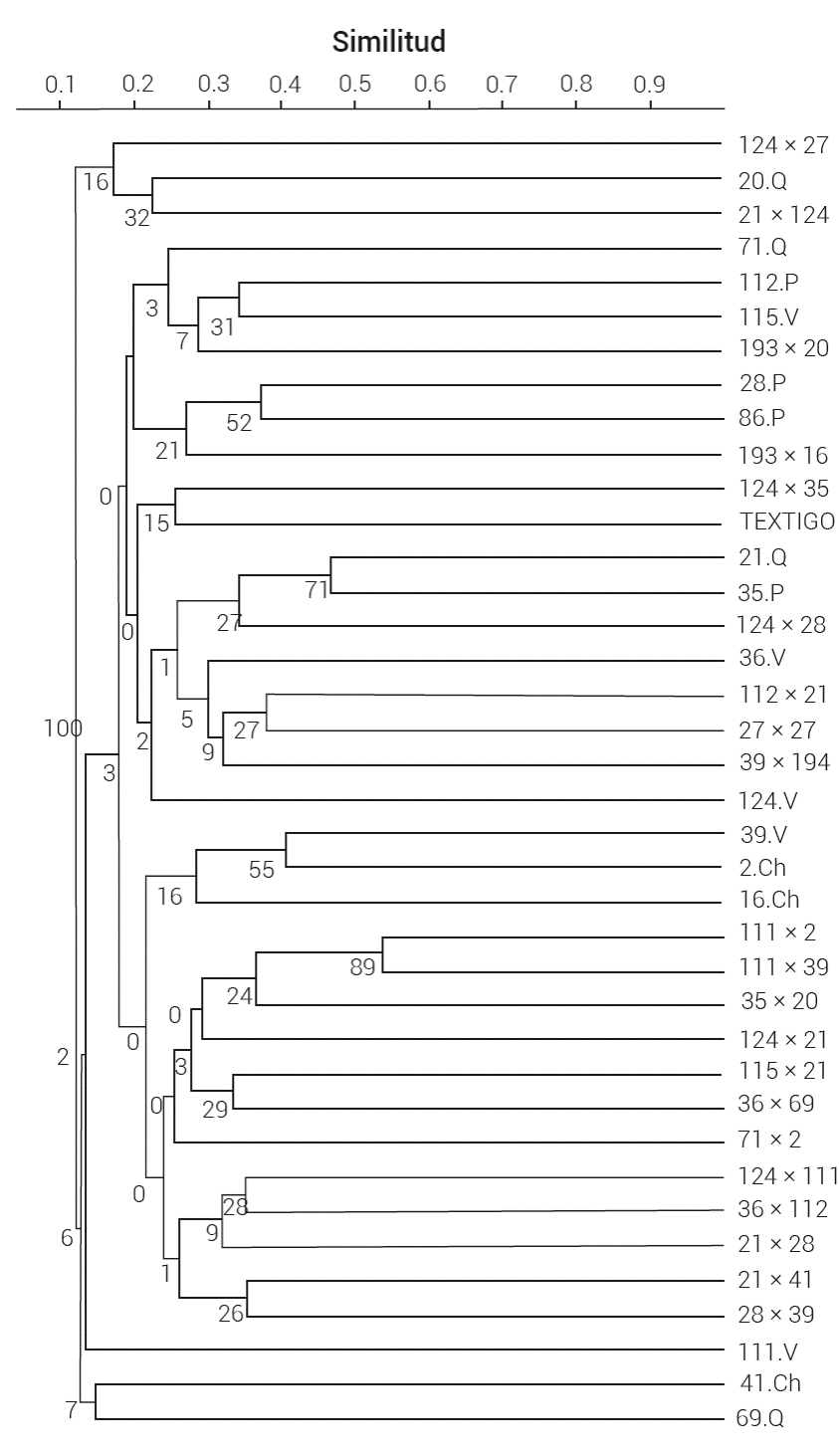
Figura 2 Dendrograma de los 21 híbridos de V. planifolia, 16 genotipos parentales y un testigo con base en las distancias genéticas de Jaccard y el método de agrupamiento UPGMA. La evaluación de cada agrupamiento está basada en 1000 repeticiones con la técnica de remuestreo bootstrap. Q: Quintana Roo, V: Veracruz, P: Puebla, Ch: Chiapas.
Estructura genética en vainilla
Con base en el estadístico ΔK, el número de grupos más probable fue K = 2; dicho resultado fue consistente con el valor encontrado por las pruebas de Gap (Tibshirani et al., 2009) y Elbow (Thorndike, 1953) del análisis de componentes principales. El primer grupo se conformó por los 16 progenitores y 11 híbridos. Cuando se usó un valor de ancestría al primer grupo mayor de 0.95, el subgrupo se conformó exclusivamente por las accesiones parentales de los estados de Quintana Roo, Puebla, Veracruz y Chiapas, México (Figura 3), lo que evidencia la existencia de un flujo genético en tales poblaciones, debido al intercambio de material biológico para el establecimiento de huertos comerciales de vainilla entre los estados de Veracruz, Puebla, Quintana Roo y Chiapas, causando que las poblaciones compartan una alta cantidad de alelos y que existan pocas diferencias alélicas entre las accesiones parentales.
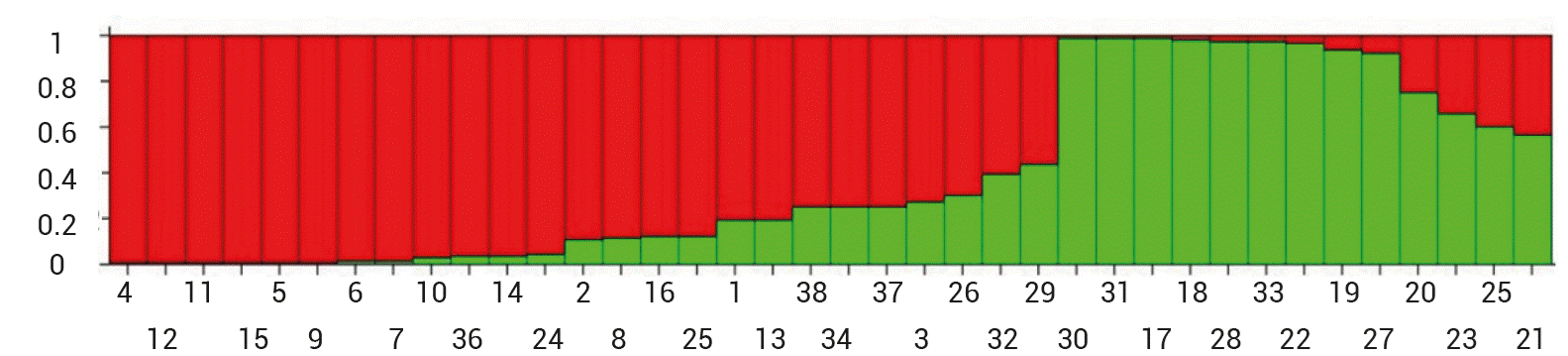
Figura 3 Estructura genética de los 38 genotipos de V. planifolia obtenidos por el análisis de agrupamiento bayesiano. Cada color representa la proporción de la probabilidad de pertenecer a cada grupo. 1-16: parentales, 17-38: híbridos.
El clon comercial utilizado como testigo en este estudio, y que es el más usado por los productores de vainilla en México, tuvo un grado de ancestría al grupo 1 de 0.75, lo cual denota un alto grado de similitud con los progenitores de vainilla. El segundo grupo se constituyó por 13 híbridos, de los cuales nueve tuvieron un grado de ancestría al grupo 2 mayor de 0.9. Tales híbridos fueron 21 × 28, 112 × 21, 124 × 21, 21 × 41, 36 × 69, 111 × 39, 111 × 2, 28 × 39 y 35 × 20, resultados que sugieren que dichos híbridos pueden contener combinaciones genéticas favorables para la producción de vainilla.
Aun cuando la variabilidad de V. planifolia pudiera ser baja en las poblaciones estudiadas, un arreglo similar fue reportado por Ramos-Castellá et al. (2017), quienes a pesar de estudiar cuatro especies de vainilla (V. odorata, V. pompona, V. insignis y V. planifolia) reportaron que el análisis bayesiano detectó dos grupos, el primero formado por V. odorata, V. pompona y V. insignis, y el segundo por todas las accesiones pertenecientes a V. planifolia, lo que confirma que existe un parecido genético entre los individuos, y por ende, una reducida variabilidad genética en V. planifolia.
Incremento de la diversidad en vainilla
Cuando el AMOVA se llevó a cabo considerando sólo los genotipos parentales, el 100 % de la varianza genética se encontró dentro de las poblaciones, lo que sugiere que la variación entre las poblaciones de los estados de Veracruz, Puebla, Quintana Roo y Chiapas, México fue muy baja (Cuadro 3); además, las pruebas de Gap (Tibshirani et al., 2009) y Elbow (Thorndike, 1953) también lo confirmaron, al mostrar que existía un solo grupo; sin embargo, la inclusión de los híbridos en el análisis genético incrementó la diversidad poblacional, ya que las dos pruebas anteriores detectaron dos grupos; igualmente, hubo un 3 % en la diversidad entre poblaciones. Adicionalmente, se observó que el grupo con el mayor porcentaje de loci polimórficos estuvo compuesto por los híbridos. Todos estos datos demuestran que la hibridación entre individuos dentro y entre poblaciones de diferente origen geográfico es un mecanismo eficiente para incrementar la diversidad génica en el cultivo de vainilla, por lo que es conveniente seguir practicando hibridación intraespecífica, y de ser posible, interespecífica, considerando que plantas de diferente origen presentan características genéticas diferentes que pueden ser aprovechadas para la obtención de materiales con mayor potencial agronómico.
Conclusiones
La variación molecular dentro de poblaciones es mayor que entre poblaciones, lo cual implica la necesidad de conservar más individuos de cada población. La hibridación entre individuos de diferente origen es una herramienta factible para incrementar la diversidad entre poblaciones, misma que puede ser usada en un programa de mejoramiento genético.

