











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCTION
The geological history of the Yucatán Peninsula has resulted in the creation of one of the biggest underground rivers in the world. With a surface area of approximately 165,000 km2 (Bauer-Gottwein et al. 2011), the limestone platform of the peninsula allows water to drip down and accumulate underground, creating water-rock interactions that result in the dissolution of carbonate rocks, evaporation, precipitation, and areas where salt and freshwater mix (Perry et al. 2009). These specific chemical conditions have allowed the formation of a flooded underground anchialine cave system approximately 330 km in length (Calderón-Gutiérrez et al. 2017) that is comparable to the sea caves of the Mediterranean that host high biodiversity, including that of sponges (Gerovasileiou and Voultsiadou 2012). However, distinct environmental conditions characterize the anchialine cave system of the Yucatán Peninsula, including light limitation, pressure and temperature variations, varying levels of dissolved organic matter, and fluctuating oxygen availability.
The conditions of the caves of the Yucatán Peninsula are so peculiar that they have generated a high degree of specialization in cave-dwelling organisms, resulting in notable endemism (Calderón-Gutiérrez et al. 2017). Indeed, a variety of endemic sponges, including novel species, have been described for 2 caves belonging to this system (Gómez and Calderón-Gutiérrez 2020), but none have been described in the caves on the mainland side of the peninsula. Sponges are sessile organisms that can filter 0.002 to 0.84 cm3·s-1 of seawater per 1 cm3 of tissue (Thakur et al. 2005). The filtered water supplies the sponges with oxygen, nutrients, and diverse microbial communities that include bacteria and archaea, which can come to constitute more than 50% of their biomass (Hentschel et al. 2003, Radjasa et al. 2011, Schmitt et al. 2012).
Previous studies have investigated the diversity of microbial communities from marine sponges using culture-independent and culture-based approaches. Moreover, 16S rRNA metabarcoding has allowed for the exploration of microbial diversity in sponge species from different environments, providing insights and new information on the importance of multiple factors, such as host species, environmental drivers, and biogeographical distributions, in shaping the composition and structure of the microbial communities of sponges (Schmitt et al. 2012, Kennedy et al. 2014, Moitinho-Silva et al. 2017, Yang et al. 2019, Busch et al. 2022). Studies using culture-based methods have demonstrated that strains isolated from sponges exhibit various biological activities, including the production of natural products with antimicrobial properties (Jayatilake et al. 1996, Su et al. 2014, Santos et al. 2015, Bennur et al. 2016). The importance of sponge-associated microorganisms lies in their contributions to the chemical defenses of their hosts against predators, competitors, and pathogens (Deshpande and Thakur 2020). Importantly, symbiotic microbes in sponges facilitate host growth in otherwise unattainable niches by recycling different sources of dissolved organic matter and making limiting nutrients available (Freeman et al. 2020).
As part of an ongoing exploration of the microbial diversity present in the anchialine cave system of the Yucatán Peninsula, we present the first description of the microbial community associated with a previously undescribed anchialine cave sponge in Xcalak, Quintana Roo, and show the potential of these associated microbes as sources of bioactive natural products. In addition, the present study represents the first effort to ecologically and biotechnologically characterize a sponge from an anchialine ecosystem, which is currently experiencing increases in human activity due to the expansion of tourism and urban development in the region.
MATERIALS AND METHODS
Sample collection, 16S rRNA sequencing, and sponge species identification
Sponge specimens were collected via scuba diving at a depth of ~12 m inside an anchialine cave (i.e., Xcalak Cave, Cayo Judío) of the underground karst aquifer sinkhole in the Yucatán Peninsula in September 2020 (Fig. 1). Cayo Judío (18°12′10.7238″ N, -87°51′38.5806″ W) is located in the State of Quintana Roo, Mexico, and is surrounded by mangrove forests and seagrass beds. Xcalak Cave is located on the coast of Cayo Judío and is characterized by a high inflow of seawater (intrusion) and little to no freshwater during the dry season (October-April). The collection date was close to the end of the rainy season; high salinities in the water column of the cave confirmed low inputs of freshwater. A permit (PPF/DGOPA-062/21) was issued for sample collection by the Secretariat of Agriculture, Livestock, Rural Development, Fisheries, and Food (SAGARPA) of Mexico.
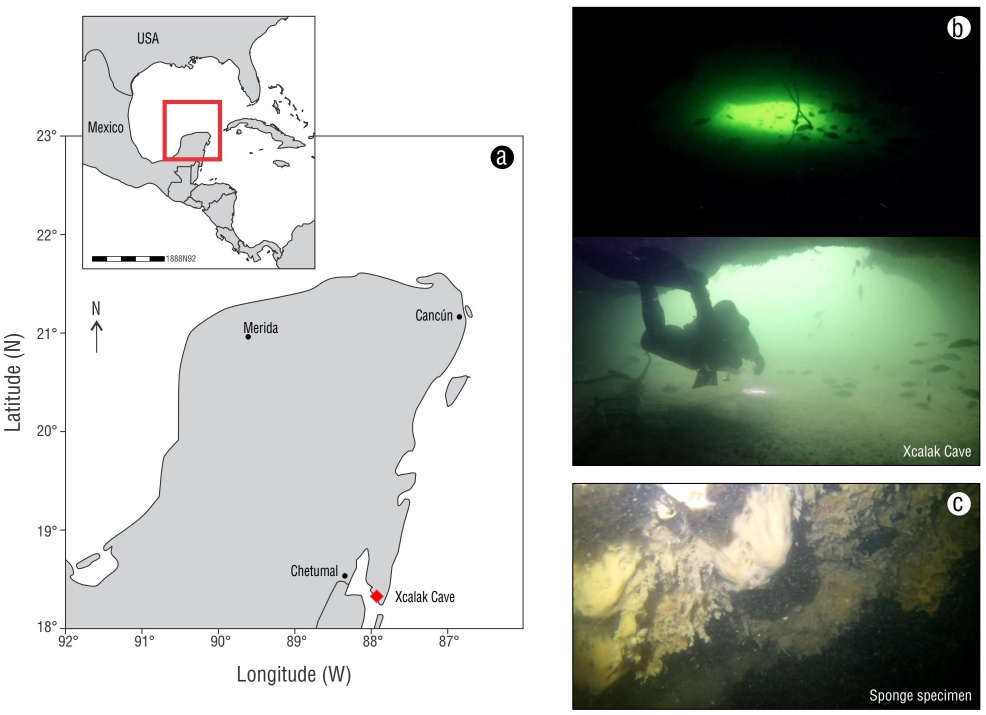
Figure 1 Study site (red diamond) in the Yucatán Peninsula (red rectangle in inset) (a), photographs of anchialine Xcalak Cave (b), and photograph of the sponge specimen (c).
The water mirror (WM) zone was determined to be the shallowest zone (1 m depth) at the entrance of the cave that was exposed to solar radiation, while the cave zone (12 m depth) was defined as the zone not exposed to solar radiation. Samples of the water column were collected in the WM and cave zones, while the sponge specimen was collected from the cave wall. After collecting the water samples in sterile bags, in situ variables, namely temperature (°C), salinity (psu), dissolved oxygen (DO, mg·L-1), specific conductance (SPC, μS·cm-1), conductance (C, μS·cm-1), electrical conductivity (mS·cm-1), total dissolved solids (TDS, mg·L-1), pH, and nitrate-N (NO3-N), were determined from on board a boat with a YSI ProQuatro handheld multiparameter (Yellow Spring Instruments, Yellow Springs, USA).
Approximately 40 cm³ of wet sponge tissue was collected in triplicate using 50-mL Falcon tubes (Corning Inc., New York, USA) and stored at 4 °C for transport to the laboratory. Once on land, the sample was preserved in 96% ethanol, photographed under a stereoscopic microscope, and identified following a traditional taxonomic approach based on the tissues and spicules (Gómez and Calderón-Gutiérrez 2020). Genomic DNA of the sponge tissue was extracted in triplicate using a ZymoBIOMICS Miniprep D4300 Kit (Zymo Research, Irvine, USA). DNA from the triplicates was pooled, and its purity and concentration were analyzed using a spectrophotometer (Nanodrop 2000, Thermo Fisher Scientific, Waltham, USA) and visualized by agarose gel electrophoresis. The DNA was stored at -20 °C for further processing.
After extraction, the genomic DNA was sent to QB3 Genomics (UC Berkeley, Berkeley, USA) (RRID:SCR_022170) for amplicon sequencing with the MiSeq 2 × 250 paired platform (Illumina, San Diego, USA). The primers 515F (5’ GTGCCAGCMGCCGCGGTAA 3’) and 806R (5’ GGACTACHVGGGTWTCTAAT 3’) were used to amplify the V4 region of the 16S rRNA gene following the protocol of Caporaso et al. (2012). The raw sequencing data of the 16S rRNA sequencing results were deposited in the Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra/) under the accession number PRJNA1118309 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1118309).
In a parallel study of the sponge microbiome conducted in our lab, the metagenome of the same tissue sample was obtained from the sequencing data generated by Novogene (Davis, USA) utilizing a NovaSeq 6000 platform (Illumina). Contigs were assembled using Metaspades v. 3.13.0 (Nurk et al. 2017) and taxonomically annotated via the basic local alignment search tool (BLASTn) against the National Center for Biotechnology Information (NCBI) nr database. The contigs that were assembled and assigned to the Eukaryota superkingdom were used to search for rRNA genes (primarily 18S rRNA) to taxonomically identify the sponge using Barrnap v. 0.9 (http://github.com/tseemann/barrnap). The rRNA sequences were aligned against a nonredundant version of the SILVA database v. 138 with an E-value <10−5 employed for taxonomic assignment. Additionally, we used another approach that involved aligning the metagenomic shotgun data (encompassing prokaryotic and eukaryotic reads) against a nonredundant version of the SILVA database v. 138 with an E-value <10−5. Sequences matching this database were considered potential rRNA gene fragments, and they were aligned against Eukarya hidden Markov models (HMMs) using SSU-ALIGN v. 0.1.1 (https://github.com/EddyRivasLab/ssu-align) to identify true sequences. Pie plots were constructed to visualize the presence and relative abundance of the genera belonging to Eukaryota and Porifera identified through both approaches (contigs and short reads) using RStudio (R Studio Team 2015). The microbial community from the sponge metagenome is still under analysis and will be reported elsewhere.
Quality filters and sequence analysis
Sequencing reads obtained from the MiSeq run were processed using Quantitative Insights into Microbial Ecology 2 (QIIME 2 v. 2020.8) (Bolyen et al. 2019). The sequences were quality-filtered with a Phred score of Q20 using the Deblur algorithm. Amplicon sequence variants (ASVs) were taxonomically assigned using the feature-classifier2 plugin implemented in QIIME2 against the SILVA SSU non-redundant database (138 release). Unassigned ASVs were submitted to a BLASTn search against the NCBI database. The ASVs assigned to Eukaryota, chloroplasts, and mitochondria were removed. A rarefaction curve was computed directly using the "diversity alpha-rarefaction” command of QIIME2 (Bolyen et al. 2019).
The most abundant taxa (from phylum to genus level) that belonged to bacteria and archaea were visualized with ‘barplot()’ in RStudio (R Studio Team 2015). A heat tree of taxonomic diversity from the sponge specimen was generated using the R package ‘metacodeR’ v. 0.2.145 (Foster et al. 2017). In these plots, the node width and color indicate the number of reads assigned to each taxon (phylum to genus level).
Isolation and molecular identification of the NCA-454 strain
The collected sponge specimen was rinsed in marine sterile water, and 1 g of tissue was cut into pieces (~1 cm³) and homogenized with a tissue tearor. Then, dilutions were performed with peptone water (0.1%). Dilutions (10-7) of peptone water were transferred to marine agar (Difco, Becton Dickinson, San Jose, USA) plates and maintained for 48 h at 25 °C; 13 halotolerant and halophilic actinobacteria were isolated. The actinobacteria strains were then grown in A1 liquid media (10 g soluble starch, 10 g yeast extract, 10 g peptone, 18 g agar, and 1 L water), and 500-µL aliquots were stored as glycerol stocks (1:1 glycerol:H2O) at -80 °C. Approximately 2 mL were used for DNA extraction using a genomic DNA isolation kit (ZymoBIOMICS DNA Miniprep Kit) following the instructions of the manufacturer (Zymo Research). To taxonomically identify strains, the 16S rRNA gene was amplified by PCR with the primers 27F (5’ AGAGTTTGATCCTGGCTCAG 3’) and 1492R (5’ GGTTACCTTGTTACGACTT 3’) (Weisburg et al. 1991). The PCR product was visualized by agarose gel electrophoresis, quantified by Nanodrop (1000) and sent to the DNA Synthesis and Sequencing Unit (USSDNA) of the Institute of Biotechnology of the National Autonomous University of Mexico (Cuernavaca, Mexico) for sequencing. A BLAST analysis was carried out via a BLASTn search in GenBank (http://www.ncbi.nlm.nih.gov) to taxonomically identify the strains. Among all isolated bacterial strains, the one identified as Nocardiopsis dassonvillei NCA-454 was selected for downstream analysis and activity assays, as it demonstrated high activity in our exploratory growth inhibition test. In addition, N. dassonvillei strains have been previously studied for their biotechnological potential (see reviews from Bennur et al. 2016 and Bhairamkar et al. 2023). The 16S rRNA sequence of N. dassonvillei strain NCA-454 was deposited in the GenBank data library under accession number OR616708.
NCA-454 extract preparation and antimicrobial susceptibility testing
Strain NCA-454 was grown in DSC-ASW media (5 g soluble starch, 10 g glucose, 5 g peptone, 5 g yeast extract, and 32 g sea salts, and 1 L water) at room temperature for 10 d to obtain sufficient cell mass for solvent extraction with 200 mL of MeOH. After extraction for 40 min in a flask, the supernatant and cell pellet were recovered by centrifugation (20 min, 6,000 rpm) and decantation. For medium (extracellular) extraction, 18 g of XAD-7 resin (amberlite) was added per liter of supernatant and incubated for 12 h on an orbital shaker (120 rpm). The resin was then filtered under vacuum, followed by extraction by maceration with 250 mL of ethyl acetate (LEtOAc) and then with 100 mL of methanol (LMeOH); this process was performed with the pellet and supernatant. This resulted in 4 different extracts (extraction fraction/extraction solvent) for Strain NCA-454: extract SA17 (supernatant/EtOAc), extract SA18 (supernatant/MeOH), extract SA19 (pellet/EtOAc), and extract SA20(pellet/MeOH).
Methicillin-resistant Staphylococcus aureus ATCC 43300 (MRSA) and methicillin-sensitive S. aureus ATCC 25913 (MSSA) were used in an in-house antimicrobial susceptibility assay to test the bioactivity of the extracts obtained from NCA-454. Briefly, an MTT solution (3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was used to calculate growth inhibition (%). Two concentrations of each crude extract of NCA-454 (SA17, SM18, PA19, and PM20) were tested (200 mg·mL-1 and 20 mg·mL-1), and an antibiotic control was used (vancomycin for MSSA and ampicillin for MRSA). High absorbances indicate low pathogen inhibition, as growing pathogens transform MTT into formazan dye. Controls for media and solvent absorbance were also used. Absorbance was determined on an iMarkTM Microplate Absorbance Reader (Bio-Rad Laboratories, Hercules, USA) at 490 and 608 nm. Percent inhibition was calculated as follows:
where blank absorbance is that of the medium, and ADMSO is the absorbance value for the solvent (DMSO).
RESULTS
Physical and chemical water properties
The WM and cave zones exhibited high salinity (34.5 ppt for both), constant temperature (31 °C), and similar values of TDS (34.13 to 34.5 mg·L-1), SPC (52,545 to 53,178 μS·cm-1), C (58,157 to 60,755 μS·cm-1), and pH (8.02 to 8.22) (Table 1). An increase from 4.3 to 6.34 in NO3 and a decrease from 4.24 to 3.77 mg·L-1 in DO were observed from the WM zone to the cave zone.
Taxonomic identification of the sponge specimen
The morphology analysis indicated that the organism was a sponge from the order Haplosclerida. Using standard identification keys based on spicule analysis, Haliclona and Xestospongia emerged as candidate genera for the sponge specimen in this study.
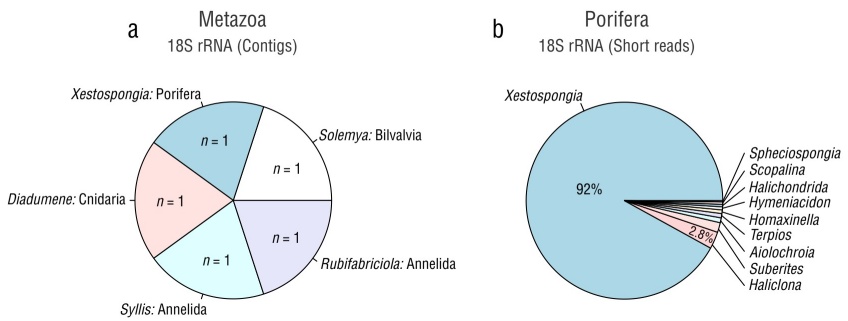
In the search for the 18S rRNA gene within the contigs assembled from the metagenomic data, 5 sequences of 18S rRNA were identified and assigned to the genera Solemya (Bivalvia, length = 1,825 bp), Rubifabriciola (Annelida, length = 503), Syllis (Annelida, length = 1,066), Diadumene (Cnidaria, length = 879 bp), and Xestospongia (Porifera, length = 2,004 bp) (Fig. 2a). Among the contigs assigned to the complete mitochondrial genome, the genus Xestospongia exhibited the highest coverage (Table S1). A different approach to analyze the 18S rRNA sequences of the phylum Porifera using short reads indicated that the genus Xestospongia accounted for the highest number of sequences (92%) (Fig. 2b), while a small proportion of sequences were assigned to the genus Haliclona (2.8%). Consequently, the sponge species utilized in this study was identified as Xestospongia sp.; identifying the sponge to the species level was not feasible due to methodological limitations.
Sponge microbial community
A total of 23,222 high-quality, filtered sequences representing 160 ASVs were obtained. A rarefaction curve of the observed ASVs showed the curve reached saturation, indicating adequate sampling of 16S rRNA sequences (Fig. S1). The 160 ASVs were assigned to different taxonomic levels: 8 phyla, 11 classes, 23 orders, 21 families, and 18 bacterial genera. In addition, a small number of ASVs were identified as Archaea (10 ASVs).
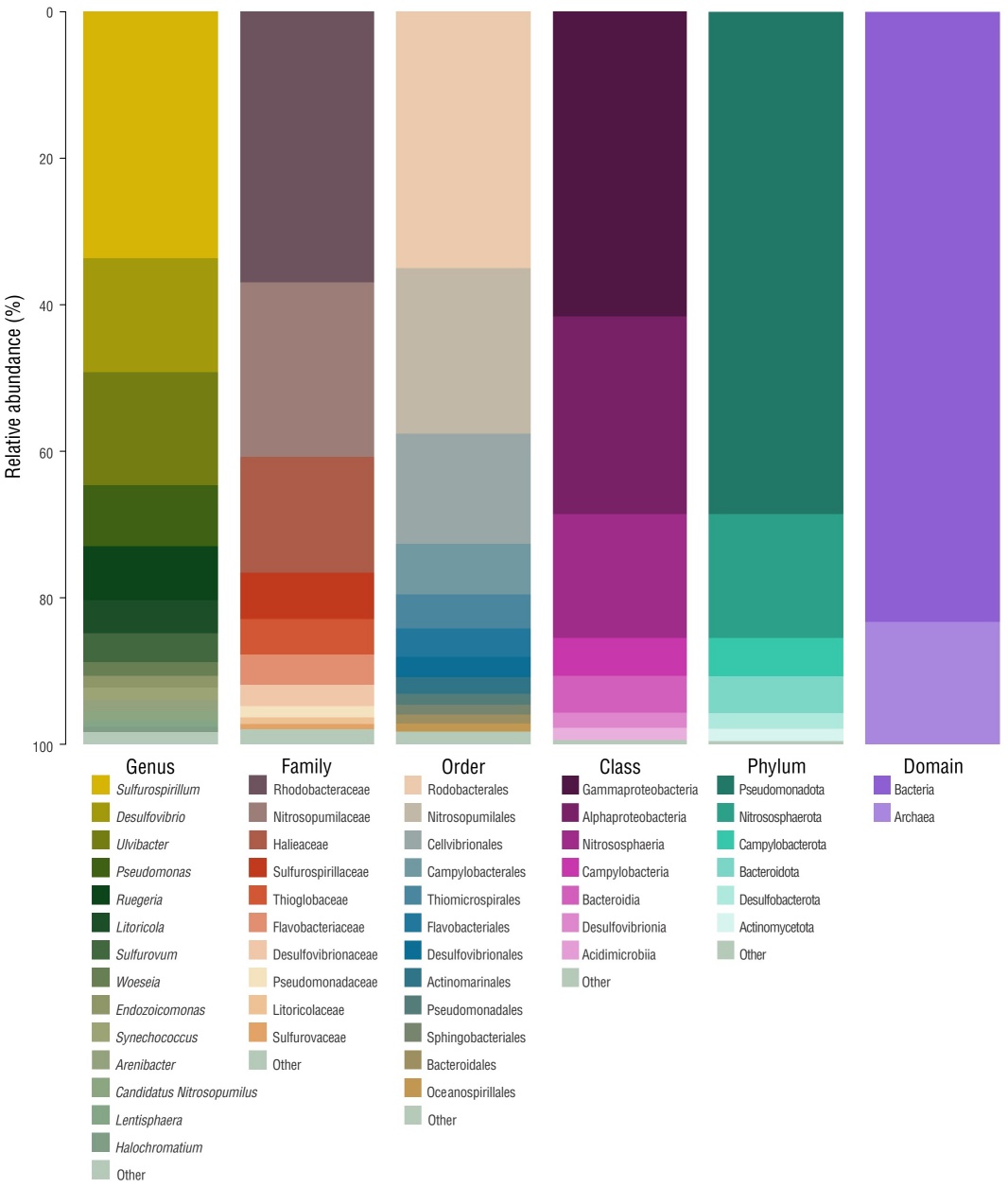
The most abundant phyla (relative abundance) were Pseudomonadota (68%), Campylobacterota (5.2%), Bacteroidota (5.1%), and Desulfobacterota (2.1%) (Fig. 3 and Fig. S2). At the class level, Gammaproteobacteria (41.6%), Alphaproteobacteria (27.0%), and Campylobacteria (5.2%) were the most dominant (Fig. 3 and Fig. S2). Rhodobacterales (35.0%), Cellvibrionales (15.0%), and Campylobacterales (6.9%) were the most abundant orders. The 3 most abundant families were Rhodobacteraceae (36.9%), Halieacea (15.8%), and Sulfurospirillaceae (6.3%) (Fig. 3 and Fig. S2). The genera Sulfurospirillum (33.6%), Desulfovibrio (15.6%), Ulvibacter (15.4%), Pseudomonas (8.4%), and Ruegeria (7.3%) were the most dominant (Fig. 3 and Fig. S2). The only genus identified for Archaea was Nitrosopumilus (Fig. 3 and Fig. S2).
NCA-454 strain identification and bioactivity
The molecular analysis of the 16S rRNA gene revealed that the strain (NCA-454) recovered from the sponge Xestospongia sp. was closely related to N. dassonvillei (100% sequence identity, E-value of 0.0) and belonged to the phylum Actinobacteria. The minimum inhibitory concentrations obtained from the antibiotic assay employing the crude extracts were 12.6 mg·mL-1 for MRSA and 4 mg·mL-1 for MSSA (Table S2). Crude extracts with a concentration of 200 μg·mL-1 showed the highest inhibition (%) against MSSA (Table 2).
DISCUSSION
Microbial diversity of the Xestospongia sp. microbiome
For the first time, we describe the microbial community associated with a Xestospongia sponge species collected from an anchialine cave in the Yucatán Peninsula. Studies on sponge microbiomes have been conducted with diverse seawater sponge species (Schmitt et al. 2012, Kennedy et al. 2014, Moitinho-Silva et al. 2017, Yang et al. 2019), including some sponges of the genus Xestospongia (Lesser et al. 2016, Thomas et al. 2016, Hayami et al. 2023). Similar microbial diversity at the ASV level has been observed in other sponges, such as Xestospongia mutua (Indraningrat et al. 2022) and Xestospongia testudinaria (Hayami et al. 2023), although both studies employed a larger number of samples than this study.
We found that Pseudomonadota, Nitrososphaerota, Campylobacterota, Bacteroidota, and Desulfobacterota were the dominant prokaryotic phyla in the microbiome of the sponge specimen. Some of these phyla (Pseudomonadota, Nitrososphaerota, and Bacteroidota) have been reported as typical microorganisms in the microbiomes of different marine sponge species (Thomas et al. 2016, Leal et al. 2022), including 4 species of genus Xestospongia found in different collection sites (Thomas et al. 2016) and X. mutua collected from the Caribbean basin (Lesser et al. 2016) and Curaçao Caribbean (Indraningrat et al. 2022). Certain phyla have been widely reported as abundant and associated with marine sponges (e.g., Cyanobacteria) while contributing to holobiont metabolism through photosynthesis. However, a low abundance of Cyanobacteria was observed in the Xestospongia sp. sample from Xcalak Cave. This could be attributed to local environmental conditions, including the lack of sunlight within the cave zone, which may have limited the relative abundance of this bacterial group within the sponge (Jahn et al. 2018).
At the genus level, Sulfurospirillum was the most abundant in the microbial community of the Xestospongia sp. from Xcalak Cave, followed by Desulfovibrio. The genus Sulfurospirillum has been associated with several environments, including sediments and groundwaters rich in sulfur compounds (Goris and Gabriele 2016). Desulfovibrio has been reported as the most dominant genus in 3 sponge species (Astrosclera willeyana, Dysidea arenaria, and Arenosclera heroni) from the South China Sea (Zhang et al. 2015). Like Sulfurospirillum, this genus has been described in facultative sulfur-reducing bacteria (SRB) that use elemental sulfur as a substrate for respiration in the absence of other possible electron acceptors (oxygen, nitrate, sulfite, or sulfate) (Fauque 1994).
The genes and metabolic pathways associated with sulfur cycling have been reported in studies of the microbiome diversity and metabolic capacity of marine sponges by metagenomic approaches (Tian et al. 2016, Lesser et al. 2022). The dominance of SRB in Xestospongia sp. suggests that an environment rich in sulfur compounds is present in Xcalak Cave, where sulfate reduction is the predominant role of SRB in the microbiome, which maintains the endosymbiotic sulfur cycle (Hoffmann et al. 2005). Furthermore, high levels of sulfate have been reported for the coastal caves of the Yucatán anchialine cave system (Schmitter-Soto et al. 2002, Suárez-Moo et al. 2022), which also supports the role of SRB in environmental biogeochemical cycles in these coastal caves.
Members of the microbial communities associated with marine sponges have been reported to exhibit various biological properties, including antimicrobial, anticancer, anti-inflammatory, antifungal, or antiviral activity (Brinkmann et al. 2017). Currently, 140 new structures, such as sesquiterpenes, polyketides, peptides, and alkaloids, have been discovered in sponge-associated microorganisms (Li et al. 2023). In this study, the genus Pseudomonas was of the most abundant genus in the 16S rRNA metabarcoding data. Previous studies of activity assays in Pseudomonas sp. strains isolated from different sponge species have reported the presence of different metabolites that inhibit the growth of microbial pathogenic strains (Brinkmann et al. 2017). Phenazine-1-carboxylic acid, obtained from Pseudomonas aeruginosa isolated from the sponge Isodictya setifera, inhibited the growth of pathogenic strains of S. aureus (Jayatilake et al. 1996). Santos et al. (2015) reported antimicrobial activity in bacterial extracts from Pseudomonas fluorescens strains against the indicator strain S. aureus ATCC 29213. The cyclic peptide diketopiperazine was isolated from these extracts and showed bactericidal activity against S. aureus and P. aeruginosa strains (Santos et al. 2015). The genus Ruegeria ranked fifth in terms of the abundance of 16S rRNA sequences, and different Ruegeria species have shown antimicrobial activity against human-pathogenic bacteria such as S. aureus, Salmonella enterica, and Candida glabrata (Almeida et al. 2023).
The only genus identified in Archaea was Nitrosopumilus. This genus has been described to belong to the ammonia- oxidizing group of archaea (AOA) and has been found to exhibit metabolic functions such as transporting phosphorus and metals from the environment (Suárez-Moo et al. 2024). Adaptations to various groundwater, geothermal, terrestrial, and marine habitats have been observed in Nitrosopumilus (Zheng et al. 2024). Furthermore, the presence of Nitrosopumilus has been reported in various hosts (Nakagawa et al. 2021), including sponges (Feng et al. 2016, Haber et al. 2021), and its presence has been associated with geochemical processes like nitrification (Feng et al. 2016) and the protection of host cells from ammonia toxicity. Furthermore, analyses of the exometabolome of cultures of strain SCM1 from the marine Nitrosopumilus maritimus showed that this AOA has the ability to biosynthesize cobalamin; genome mining of this AOA strain detected genes involved in the biosynthesis of metabolites, such as agmatidine and medicagenate (Law et al. 2021), demonstrating the high biotechnological potential for this genus. This potential could be further explored in AOA strains of sponges from the anchialine caves of the Yucatán Peninsula.
We recognize the limitations of partially sequencing the 16S rRNA gene in a microbiome study, as well as using only one sponge specimen. No statistical tests were conducted in this study due to the lack of replicate sponge samples in the metabarcoding analysis because this specimen was the only one found in the cave. Further investigations should involve a larger number of sponge samples and explore factors such as host species and the environmental variables influencing the microbial community. The first insights into the microbiome diversity of the sponge provided by this study will support the generation of novel hypotheses of roles of these microbes in this poorly explored environment. Future research should focus on studying the microbial community of the sponge by metagenomic approaches, including shotgun and long reads with Pacbio or Nanopore, using a higher number of samples. Nevertheless, we did not find more than one individual sponge of this species in Xcalak Cave during sampling (Efrain Chavez and Luis Liévano, pers. comm.).
Antibiotic activity of the Nocardiopsis dassonvillei NCA-454 strain
Strain NCA-454 was isolated using sponge tissue as inoculum; its 16S rRNA sequence showed high similarity to N. dassonvillei (KU306741.1/KF543091.1/MN108027.2/MN371461.1 with 99% sequence identity). The genus Nocardiopsis is composed of halotolerant and halophilic species, and members of this genus have been frequently isolated from areas with high salt concentrations (Bennur et al. 2016) like the anchialine Xcalak Cave in Cayo Judío. The crude extracts obtained from N. dassonvillei NCA-454 inhibited the growth of MRSA and MSSA. Antimicrobial activity against multidrug-resistant pathogens belonging to one gram-positive and 6 gram-negative bacteria has been observed for the cell-free supernatant of several N. dassonvillei strains isolated from different marine sponges (Selvin et al. 2009). Another N. dassonvillei strain has shown antimicrobial activity against Vibrio dibolicus, Vibrio parahaemolyticus, and Bacillus subtilis (Su et al. 2014). Chemical analyses of other Nocardiopsis extracts have uncovered the production of secondary metabolites, including polyketides, phenzines, quinoline alkaloids, terphenyls, proteins, thiopeptides, and amines (Bennur et al. 2016), all of which could be responsible for the observed inhibition of pathogenic MRSA and MSSA by N. dassonvillei NCA-454 in this study. Furthermore, whole genome sequencing of Nocardiopsis sp. TP-A0876 highlighted the presence of 3 polyketide synthases (Komaki et al. 2014), while genome mining of Nocardiopsis sp. RACA4 isolated from a Red Sea nudibranch showed a variety of secondary metabolites, namely polyketides, terpenes, and nonribosomal peptides (Elfeky et al. 2023). The bioactivity observed in N. dassonvillei NCA-454 crude extracts stresses the importance of efforts to discover secondary metabolites from cultivable bacteria in the microbiomes of sponges from diverse and novel environments such as the anchialine cave system in the Yucatán Peninsula.
CONCLUSIONS
This study represents the first effort to characterize the microbial diversity and biotechnological potential of bacteria isolated from the sponge Xestospongia sp. collected in an anchialine cave in the Yucatán Peninsula. Our cultivation-independent analysis identified several bacterial members known for their ability to produce natural products and participate in biogeochemical cycles. Furthermore, this study highlights the biotechnological significance of exploring novel environments in the search for bacterial strains harboring bioactive natural products. Future research should focus on the function (metagenomic) and gene expression patterns (metatranscriptomic) associated with the microbial community from the anchialine cave sponge that we report here.

