











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Prune Belly (SPB) es una enfermedad congénita rara caracterizada por una tríada que consiste en deficiencia de la musculatura de la pared abdominal, criptorquidia bilateral y anomalías del tracto urinario (hidroureteronefrosis, megauréter). Afecta predominantemente a los varones con una incidencia de 3.4-3.8 por 100,000 nacimientos de hombres vivos en los EEUU; este síndrome se presenta en menos del 5% en mujeres.1 Los pacientes con defectos de pared abdominal unilateral, o del sexo femenino se clasifican como síndrome pseudo Prune Belly, o bien, síndrome Prune Belly incompleto o parcial.2,3
Frolich reportó el SPB por primera vez en 1839 y en 1950 Eagle y Barret informaron sobre nueve casos y los describieron como “síndrome de Eagle-Barret”; sin embargo, Osler acuñó el término “síndrome de Prune Belly”, tomando su nombre de los cambios en la pared abdominal.4,5 Existen otros sinónimos, como “síndrome de tríada”, “síndrome de abdomen en ciruela pasa” y “síndrome de deficiencia de musculatura abdominal”.4
En la actualidad, existen múltiples teorías acerca de su patogénesis. Algunas incluyen el origen genético heterogéneo, con la implicación de diversas proteínas de contractilidad muscular visceral o de alteraciones de los receptores muscarínicos colinérgicos M3,6,7 o bien, que el modo de herencia está ligado al cromosoma X.8,9 Otra propuesta describe que hay alteración en el desarrollo del mesodermo, lo cual explicaría las alteraciones morfológicas a nivel genitourinario, gastrointestinal y de otros sistemas.10
El diagnóstico puede realizarse durante la gestación, a través de una ecografía obstétrica con evidencia de contornos irregulares en el abdomen asociado con un aumento de su circunferencia, anomalías del tracto urinario, oligohidramnios, ascitis fetal y uraco permeable. Sin embargo, este síndrome se diagnostica con mayor frecuencia al nacimiento por la particular apariencia de la pared abdominal.11 Se han propuesto diferentes opciones de tratamiento de esta compleja condición, que requiere la participación de especialistas con un enfoque interdisciplinario.4 Ante la escasa literatura sobre esta condición en pacientes de sexo femenino, se presenta el caso de una adolescente con diagnóstico de SPB desde el nacimiento, que ha requerido múltiples intervenciones quirúrgicas y manejo multidisciplinario.
Presentación del caso
Mujer de 17 años con síndrome de pseudo Prune Belly, quien asiste a manejo en una institución desde su nacimiento. El diagnóstico se realizó en etapa neonatal por la observación de defectos en la musculatura abdominal con piel laxa y arrugada, y después con la identificación en ecografía renal y de vías urinarias de dilatación del tracto urinario superior del lado derecho.
La paciente fue producto del primer embarazo, con controles prenatales adecuados, sin casos similares en la familia. Nació a las 28 semanas de gestación de manera prematura. Ha requerido múltiples hospitalizaciones por procedimientos quirúrgicos e infecciones urinarias. También se ha detectado estreñimiento crónico funcional, escoliosis toracolumbar y discapacidad cognitiva leve.
La primera intervención quirúrgica fue en el periodo neonatal por malformación anorrectal asociada a anomalías del tracto urinario; se realizó vesicostomía, colostomía y anorrectoplastia sagital posterior. La colostomía se cerró a los tres años. A los 12 años requirió realizar vaginoplastia, neovejiga derivada de íleo y colon, apendicovesicostomía (canal Mitrofanoff) por urología pediátrica y cirugía plástica.
Se ha mantenido en vigilancia por nefrología pediátrica por diferentes episodios de infección de vías urinarias (IVU), siendo la última a los 15 años. A los 11 años presentó deterioro de tasa de filtración glomerular (TFG) a 71 mL/min/1.73 con gammagrafía renal DMSA, con el cual también se evidenció riñón izquierdo hipoplásico y contribución de 13% de la TFG. El riñón derecho con hidronefrosis y contribución de 87%. En último control con ecografía de vías urinarias, se observó hidronefrosis derecha grave (20 mm) (Figura 1A) y atrofia renal izquierda (Figura 1B).
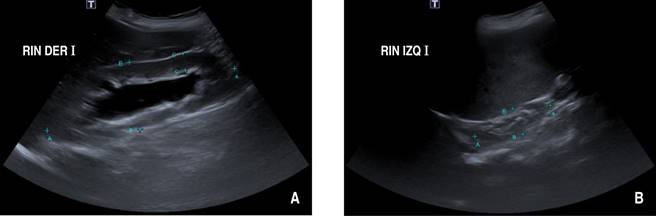
Figura 1: Ecografía renal y de vías urinarias. A) Hidronefrosis del riñón derecho. B) Atrofia renal izquierda.
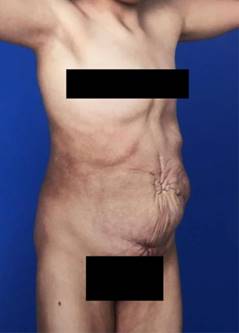
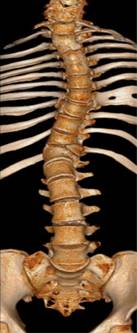
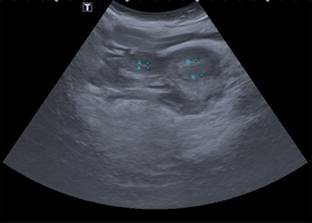
A la exploración física actual, el abdomen es prominente y globoso, con dibujo de asas intestinales en la piel (Figura 2), y existe escoliosis toracolumbar (Figura 3). Además, se encuentra en seguimiento por ginecología pediátrica porque tiene genitales externos poco desarrollados, útero didelfo (Figura 4) y presenta hemorragia uterina disfuncional. Su manejo es con terapia de reemplazo hormonal y dilataciones vaginales.
Por último, conviene señalar que no se le realizaron estudios moleculares y no fue valorada por el servicio de Genética durante los primeros años de vida.
Discusión
El síndrome de pseudo Prune Belly o Prune-Belly incompleto y el SPB se considera que pertenecen a un solo grupo de defectos congénitos con diferentes grados de afectación y severidad de las manifestaciones. Sin embargo, a la luz del conocimiento, el término de pseudo Prune Belly se encuentra en desuso.
Las principales anormalidades descritas de esta condición son en la musculatura abdominal, renal y de vías urinarias; sin embargo, entre 65 y 73% de los casos presentan manifestaciones no urológicas, incluidas anomalías gastrointestinales, ortopédicas, cardiacas, pulmonares y asociaciones con el grupo VACTERL (defectos vertebrales, atresia anal, defectos cardiacos, fístula traqueoesofágica, alteraciones renales y alteraciones en extremidades).12 En la presentación en mujeres se describen también malformaciones en genitales.
Las anormalidades renales representan el mayor determinante de supervivencia; alrededor del 30% de los pacientes presentan IVU o falla renal durante los primeros dos años de vida.13 Respecto a las manifestaciones del tracto urinario, a menudo existe dilatación vesical, hidroureteronefrosis y vejiga hipotónica, con escasa capacidad de acomodación por insuficiencia en la contracción del detrusor, lo que ocasiona reflujo vesicoureteral y aumento de residuo postmiccional, el cual es peor en sujetos con válvula uretral posterior, lo que propicia infecciones urinarias y enfermedad renal crónica.1,5,14
El SPB tiene una alta morbilidad y mortalidad, de 20 a 30% mueren en los primeros meses de vida. La mayoría mueren debido a la gran cantidad de alteraciones orgánicas o por las complicaciones inherentes a la prematuridad, lo cual está presente en 75% de los casos.1,15 En el caso descrito, presenta las típicas manifestaciones descritas de la pared abdominal, además de anomalías en el tracto urinario y alteraciones ginecológicas (genitales externos subdesarrollados, útero didelfo) malformación anorrectal, así como escoliosis.
Es escasa la literatura científica acerca de mujeres con SPB, en particular de adolescentes o adultas, ya que la mayoría son reportes de casos de recién nacidos, lo que no permite conocer la evolución a largo plazo. Reinberg y colaboradores describieron siete mujeres con SPB y su seguimiento desde el nacimiento, evidenciando falla renal en dos pacientes de las cuatro sobrevivientes, quienes requirieron trasplante renal a los 13 y 14 años respectivamente, de manera exitosa.16
Se han considerado múltiples teorías sobre su etiología, siendo las más aceptadas las alteraciones en la embriogénesis como obstrucción al tracto de salida vesical, secundaria a uretra displásica o disfuncional por canalización tardía de la uretra,4,12 disgenesia del saco vitelino y trastorno del desarrollo mesodérmico, entre la sexta y décima semana de gestación, lo que explicaría las alteraciones morfológicas en los diferentes órganos y sistemas.3,5,10,12 Se han estudiado bases genéticas y mecanismos de herencia para el síndrome y se han propuesto algunos genes implicados en la fisiopatología, como la alteración de los receptores muscarínicos colinérgicos M3 o CHRM3, en el cromosoma 1q43 con herencia autosómica recesiva, el cual favorece el desarrollo del epitelio renal y el músculo de la vejiga. Esto se observó por primera vez en una familia consanguínea con SPB que tenía una mutación homocigótica en dicho gen.7,12,17
También se ha estudiado la participación del cromosoma X. En un estudio de Iqbal y colaboradores se identificaron dos pacientes con desequilibrio en el cromosoma X, y uno con duplicación del gen AGTR2 ubicado en dicho cromosoma, el cual está implicado en el desarrollo del tracto urinario.8 En otro estudio se identificaron tres mutaciones del gen FLNA, implicado en la producción de la proteína filamina A, la cual regula la reticulación de actina que funciona en las células del músculo liso.9 Además, se han propuesto teorías que implican alteraciones en diversas proteínas de contractilidad muscular visceral como resultado de mutaciones genéticas, incluyendo los genes ACTA2, ACTG2, MYH11, MYLK, MYOCD y HNF1B.6
El diagnóstico precoz permite un tratamiento oportuno. La descompresión vesical intrauterina se ha descrito mediante la creación de una derivación vesicoamniótica. El diagnóstico prenatal es posible desde las semanas 11 y 14, pero en la mayoría de los casos se realiza en el segundo trimestre. En ultrasonido se observa megavejiga, riñones hiperecoicos, hidrouréteres e hidronefrosis bilateral.4 En nuestro caso, el diagnóstico fue en el momento del nacimiento por los hallazgos al examen físico y las alteraciones del tracto urinario evidenciadas por ecografía.
El SPB tiene varias manifestaciones clínicas y una gravedad muy variable, por lo que el tratamiento debe dirigirse a las necesidades específicas del paciente y su calidad de vida.1 En la mayoría de los pacientes se realiza injerto de la pared abdominal con una supervivencia del injerto de uno a cinco años de 66.7%.18 Los pacientes generalmente se someten a una o dos cirugías por año durante la infancia; sin embargo, no se ha establecido el mejor momento para este procedimiento. Algunos autores proponen hacerlo temprano para evitar la dilatación progresiva del sistema urinario, pero otros argumentan que es preferible un tratamiento conservador inicial.1,4,19

