











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las acidemias orgánicas (AO) son errores innatos del metabolismo causados por deficiencias en proteínas involucradas en las vías metabólicas intermediarias de los carbohidratos, aminoácidos y ácidos grasos que producen acumulación de ácidos orgánicos en diferentes tejidos y su excreción elevada en orina.1 Aunque la prevalencia e incidencia de las AO son bajas,2 hay regiones del mundo con mayor frecuencia de presentación, como en India y otros países del Oriente Medio. En Inglaterra se estima una incidencia de 3.7 casos por cada 100,000 recién nacidos vivos.1
Con la implementación en varios países del tamizaje metabólico neonatal se ha observado un aumento de los casos detectados,3 lo cual sugiere que las AO se han subdiagnosticado, particularmente en países donde no se realiza esta prueba. Cabe resaltar que sin un diagnóstico y manejo oportuno en las primeras 24 a 48 horas que inician los síntomas, el pronóstico se ensombrece con mayor probabilidad de complicaciones neurológicas a largo plazo, progresión a coma y muerte.4,5
La acidemia isovalérica (IVA) es una de las AO clásicas. Se presenta como consecuencia de la deficiencia genética de la isovaleril-CoA-deshidrogenasa, la cual es responsable de catalizar el tercer paso del metabolismo de la leucina. El bloqueo de la vía genera la acumulación de metabolitos tóxicos derivados del isovaleril-CoA como el ácido valérico, el ácido 3-hidroxi-isovalérico, la isovalerilcarnitina y la isovalerilglicina (Figura 1).6
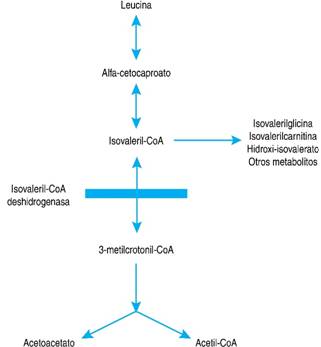
Figura 1: Vía metabólica de la leucina. Con una barra se indica el sitio del bloqueo metabólico en IVA.
El presente reporte corresponde a un paciente en quien se realizó el diagnóstico de IVA en las primeras 48 horas en que se presentaron los síntomas.
Presentación del caso
Recién nacido masculino, producto de la primera gestación de padres jóvenes consanguíneos (segundo grado) sin otros antecedentes perinatales de relevancia. Durante los primeros cinco días de vida el paciente se encontró sin problemas; al sexto día, mostró irritabilidad, somnolencia y succión débil. Fue llevado para atención médica y se hospitaliza en la Unidad de Cuidados Neonatales por sospecha de sepsis neonatal. Al examen físico, el peso, talla y perímetro cefálico eran adecuados para la edad; sin embargo, se observó una pérdida de peso mayor de lo esperado en la primera semana de vida, signos vitales normales, sin alteraciones en el patrón respiratorio, cardiaco o síntomas gastrointestinales. No obstante, estaba somnoliento e hipoactivo con reflejos primitivos disminuidos e incompletos. En los exámenes de laboratorio se evidenció hiperglucemia (182 mg/dL) y acidosis metabólica con anión GAP elevado (no se dispone de datos numéricos), ante estos hallazgos se inició manejo antibiótico con gentamicina y ampicilina, soporte con líquidos endovenosos y reposición de bicarbonato.
La respuesta clínica no fue satisfactoria; continuaba somnoliento e hipoactivo. En la determinación de amonio se observó un valor de 292 μmol/L, (límite superior: 100 μmol/L), por lo que se da manejo para hiperamonemia: se suspende la vía oral con ajuste del flujo metabólico y se indica ácido carbaglúmico. Se realizaron tres controles de amonio en las siguientes 24 horas, con valores de 292 μmol/L, 31.86 μmol/L y 31.27 μmol/L, respectivamente. Se completaron los estudios con aminoácidos cuantitativos en plasma por HPLC, cromatografía de ácidos orgánicos en orina, lactato y piruvato séricos.
Los aminoácidos cuantitativos en plasma, lactato y piruvato se reportaron dentro de límites normales, en la cromatografía de ácidos orgánicos en orina se observó elevada excreción de isovalerilglicina, ácidos 3-hidroxi-isovalérico, isovalerilglutámico y 2-hidroxi-isovalérico. Con este perfil metabólico se diagnosticó acidemia isovalérica (Figura 2), por lo que se indica fórmula especializada libre de leucina, dieta con restricción proteica y L-carnitina, como coadyuvante. Así, el paciente mostró respuesta clínica y bioquímica favorable, con amonio dentro de valores normales y corrección de la acidosis metabólica. Se egresó alerta y activo.
A los seis meses de edad, al tratamiento se adicionó L-glicina y L-leucina. Esta última la recibió por un periodo corto de tiempo, pues no se identificó una clara indicación. Se solicitó cromatografía de aminoácidos cualitativos en plasma, los cuales fueron normales. El perímetro cefálico, peso y talla se encontraban entre -2 y -3 desviaciones estándar, pero persistía con hipotonía. Sin embargo, el neurodesarrollo era acorde a la edad, logrando sedestación en trípode, prensión palmar radial de objetos de forma simétrica, balbuceo y búsqueda de interacción con sonrisas.
Discusión
La mitad de los pacientes con IVA inicia la sintomatología dentro las primeras cinco semanas de edad, la cual se caracteriza por succión débil, vómito, acidosis metabólica, cetosis, deterioro progresivo del estado de conciencia, crisis convulsivas y, de continuar, puede progresar coma y muerte. La otra mitad de los pacientes inicia los síntomas cuando son lactantes o preescolares y algunos pocos en la adolescencia. En otros países se han reportado pacientes asintomáticos detectados mediante el tamizaje metabólico neonatal;7 en Colombia este tipo de tamizaje no está disponible, por lo que sólo se detectan pacientes con manifestaciones clínicas establecidas.
Varios pacientes con algún tipo de AO, como ocurrió en el caso descrito, son diagnosticados con sepsis neonatal temprana, esto se debe principalmente a la poca especificidad de los síntomas y signos en el periodo neonatal; por lo tanto, es importante tener la sospecha clínica para el diagnóstico, prestando especial atención a los pacientes que no responden de manera adecuada al manejo convencional o en quienes predomina el compromiso neurológico.8
Al igual que en otros errores innatos del metabolismo, la IVA presenta un espectro metabólico y clínico amplio, de tal manera que el compromiso metabólico da lugar a la división en dos grandes grupos, de acuerdo a los niveles de N-isovalerilcarnitina en el tamizaje neonatal: IVA con compromiso metabólico grave y otro grupo con compromiso leve.9
El espectro de las manifestaciones puede ser de leve a grave; los pacientes con un cuadro leve tienen poca afección neurológica o en el neurodesarrollo. Para quienes tienen un cuadro clínico moderado, el compromiso neurológico es mayor y con algún grado de retraso en el neurodesarrollo, el cual puede ser reversible con el tiempo. Los pacientes con mayor gravedad tienen las manifestaciones neurológicas más importantes y con marcado retraso en el neurodesarrollo.10 El compromiso metabólico en nuestro paciente, a pesar de no disponer de los niveles de N-isovalerilcarnitina, probablemente fue severo. La cromatografía de ácidos orgánicos arrojó un perfil clásico de IVA asociado a manifestación clínica grave, dado el compromiso neurológico y la instauración temprana de los síntomas.
Como diagnósticos diferenciales se deben considerar otros errores innatos del metabolismo, que también inician con sintomatología en el periodo neonatal y presentan hiperamonemia. Es importante recordar que si la hiperamonemia se instaura en el primer día de vida, puede tratarse de hiperamonemia transitoria, en especial si es un prematuro. La hiperamonemia puede ser secundaria a deficiencia de piruvato deshidrogenasa o aciduria glutárica tipo II, principalmente en recién nacidos a término. Si los síntomas y la hiperamonemia inician después de las 24 horas de vida, como ocurrió en nuestro paciente, la presencia de acidosis metabólica debe orientar el diagnóstico hacia AO, deficiencia de piruvato carboxilasa, trastorno de la betaoxidación o acidosis láctica.11 En un estudio de 21 pacientes con IVA que habían tenido 69 episodios de descompensaciones metabólicas, el 20% presentó hiperamonemia.7 Otros hallazgos de laboratorio incluyen hipoglucemia o hiperglucemia (esto último ocurrió en el paciente que describimos), cetonuria, hipercalcemia, anemia, trombocitopenia y leucopenia.12 En nuestro paciente, en conjunto al cuadro clínico, los resultados de amonio elevado con la acidosis metabólica con anión GAP aumentado y bicarbonato bajo orientaron el enfoque diagnóstico hacia AO, puesto que son hallazgos comunes en este tipo de error innato en el metabolismo en el periodo neonatal.5
El análisis de ácidos orgánicos en orina se considera la herramienta de elección para la confirmación bioquímica de las AO. Más de 100 ácidos orgánicos son excretados en la orina en estas patologías y cada uno de ellos presenta un patrón excretorio característico, el cual permite llegar al diagnóstico bioquímico (Tabla 1).12 La cromatografía de ácidos orgánicos en orina de nuestro paciente mostró un perfil compatible con IVA por excreción elevada de isovalerilglicina, 3-hidroxi-isovalérico, ácido isovalerilglutámico y 2-hidroxi-isovalérico. Si se solicitan acilcarnitinas es posible encontrar elevación de isovalerilcarnitina (C5), la cual no es específica de IVA, ya que puede encontrarse en otros errores innatos como la aciduria metilbutírica, o también por el uso de cremas a base de ácido piválico.
Tabla 1: Perfiles urinarios típicos de las acidemias orgánicas clásicas.
| Trastorno | Deficiencia enzimática | Perfil de ácidos orgánicos |
|---|---|---|
| Acidemia isovalérica | Isovaleril-CoA deshidrogenasa | Ácido 3-hidroxi-isovalérico |
| Ácido 4-hidroxi-isovalérico | ||
| Isovalerilglicina | ||
| Isovalerilglutamato | ||
| Acidemia propiónica | Propionil-CoA carboxilasa | Ácido 3-hidroxipropiónico |
| Ácido metilcítrico | ||
| Propionilglicina | ||
| Ácido 2-metilglutacónico | ||
| Ácido 3-ceto-2-metilvalérico | ||
| Ácido 3-ceto-2-metilbutírico | ||
| Tiglilglicina | ||
| Acidemia metilmalónica | Metilmalonil-CoA mutasa | Ácido 3-hidroxipropiónico |
| Trastornos del metabolismo de la cobalamina | Ácido metilmalónico | |
| Ácido metilcítrico | ||
| Propionilglicina |
Adicionalmente, se encuentra disponible el análisis molecular de la IVA. A la fecha se han descrito cerca de 40 mutaciones asociadas con IVA, siendo la mutación c.932C>T (p.A282V) la más frecuente, ya que se observa en 47-52% de los pacientes detectados por el tamiz neonatal con un perfil bioquímico leve, lo anterior contrasta con las mutaciones heterogéneas descritas en pacientes con perfiles bioquímicos clásicos.10
El tratamiento para IVA puede ser más efectivo cuando el diagnóstico se realiza más tempranamente. La base son las dietas restrictivas de leucina y fórmulas basadas en aminoácidos libres de leucina, con lo cual se disminuye la producción de metabolitos tóxicos. Se debe recordar que el manejo nutricional debe proveer la energía, el nitrógeno, las vitaminas y los minerales necesarios para promover el anabolismo y crecimiento.7 Las fórmulas sintéticas deben aportar aproximadamente 50% de los requerimientos proteicos diarios. La deficiencia de carnitina es frecuente, por lo que se debe realizar seguimiento periódico de los niveles de carnitina libre. El manejo adyuvante con L-carnitina y L-glicina aumenta el número de metabolitos conjugados, menos tóxicos en comparación con los metabolitos libres del ácido isovalérico.7,13
El tratamiento agudo, cuando hay descompensación metabólica, incluye el manejo de la acidosis e hiperamonemia, corrección de líquidos, glucemia y electrolitos; además de procurar la adecuada perfusión y disponibilidad de oxígeno. En el caso que se presenta, el manejo con ácido carbaglúmico tuvo un rol importante para lograr estabilizar los niveles de amonio en menos de 24 horas. El seguimiento interdisciplinario debe ser individualizado, según la edad del paciente y la severidad metabólica y clínica. Debe haber evaluación periódica de gasometría, amonio y aminoácidos cuantitativos en plasma prestando mayor atención a los niveles de isoleucina, metionina, treonina y valina, cuyos niveles pueden caer abruptamente al iniciar el tratamiento, poniendo al paciente en riesgo de desarrollar manifestaciones asociadas a la depleción de éstos, de ahí que es esencial la vigilancia nutricional.14 Por último, la asesoría genética resulta relevante, en especial para la detección prenatal en caso de que se desee un nuevo embarazo.15

