











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Se define como ictiosis congénita autosómica recesiva a un fenotipo genérico de eritrodermia, piel escamosa que se presenta sobre casi toda la superficie del cuerpo al momento del nacimiento, pero sin otras manifestaciones extracutáneas.1
En la mayoría de los casos, hay un historial familiar de la enfermedad o consanguinidad. Se distinguen por su modo de herencia, características clínicas, defectos asociados y hallazgos histológicos.2
La última clasificación de consenso de ictiosis, diferencia entre dos formas principales: las no sindrómicas (que cursan con manifestaciones solamente en la piel) y las sindrómicas (que se presentan con manifestaciones en otros órganos). Entre las formas no sindrómicas, se han identificado cuatro grupos: ictiosis comunes, ictiosis autosómica recesiva congénita (ARCI, por sus siglas en inglés), ictiosis queratinopática y otras ictiosis menos comunes. De manera tradicional, el grupo de ARCI se ha dividido en dos trastornos: ictiosis lamelar (LI) y eritrodermia ictiosiforme congénita (CIE). En la clasificación más reciente, a este grupo se agregó a la ictiosis arlequín (HI).3
Se dispone de datos limitados sobre la epidemiología de las ARCI. En los Estados Unidos de Norteamérica se ha estimado una prevalencia al nacer de uno por 100,000 habitantes para LI y de uno por 200,000 habitantes para CIE. Otros estudios han informado una combinación de prevalencia para LI y CIE de uno por 200,000 a 300,000.4
Los bebés colodión nacen con una membrana tensa, brillante, translúcida u opaca que recubre todo el cuerpo y dura de días a semanas. Los tipos LI y CIE son fenotipos aparentemente distintos: LI presenta escamas color marrón oscuro, en forma de placa, sin eritrodermia; mientras que en la CIE, las escamas tienen una coloración más blanca, fina y existe eritrodermia generalizada. Las formas graves pueden presentar además ectropión, eclabio, alopecia cicatricial (que afecta el cuero cabelludo y las cejas) así como queratodermia palmar y plantar.5
El diagnóstico de ARCI no sindrómico se establece por los hallazgos de la piel al momento de nacer y en los primeros meses de vida. La biopsia de piel no es necesaria para establecer el diagnóstico de ARCI.
Nos propusimos presentar el presente caso dado que en la literatura la recurrencia de ictiosis en una misma familia es una condición poco informada.
Presentación del caso
Informamos el caso de una recién nacida (RN) femenina de término, padres originarios de Veracruz y residentes del Estado de México; negaron exposición a sustancias tóxicas o radiografías. Existía historia de ictiosis en el árbol genealógico, en familiares de tercer grado, además describieron algunos casos que se han presentado en su comunidad natal. Padres consanguíneos en cuarto grado; la primera hija tuvo diagnóstico de ictiosis y falleció tres meses después a consecuencia de sepsis, cuando se encontraba en protocolo de clasificación clínica para ictiosis sindrómica.
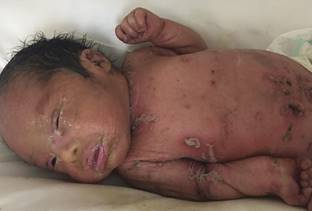
La madre tenía 17 años y acudió a control prenatal desde el primer trimestre, asistiendo a ocho consultas en el centro de salud; negó infección del tracto urinario. No se realizó ecografía obstétrica. Inició su trabajo de parto a las 37 semanas de gestación, el cual se resolvió por parto eutócico. La RN tuvo Apgar de 8 y 9, peso: 2,780 g; perímetro cefálico: 33 cm; longitud: 49 cm; perímetro del tórax: 31 cm; perímetro abdominal: 31 cm. En el momento del nacimiento se encontró una membrana constrictiva y tensa que cubría toda la superficie del cuerpo; la piel se observó eritematosa, brillante, estirada y esmaltada, con fisuras en el cuello y tronco. Se detectó ectropión del párpado y configuración de los labios en forma de “O” (Figuras 1, 2 y 3). Las uñas y el cabello eran normales; el resto de la exploración fue prácticamente normal.
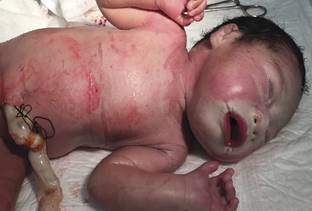
Figura 1: Piel eritematosa con fisuras en el cuello y tronco, brillante, estirada y esmaltada; configuración de los labios en forma de “O”.
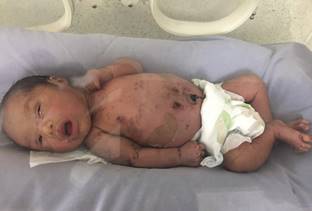
Figura 2: Bebé colodión; se observa en la incubadora con áreas de descamación en grandes capas y presencia de una pústula abdominal.
La paciente fue hospitalizada en la Unidad de Cuidados Intensivos Neonatales; se encontraba inicialmente en estado de hipotermia y luego presentó fiebre (39-40 °C). Se colocó catéter umbilical para su manejo hídrico. Fue tratada de forma estéril y se le mantuvo en una incubadora con humidificador para mantener la termorregulación. El estudio de sangre de rutina reveló fórmula roja sin alteraciones y leucocitosis; las pruebas de función renal estaban ligeramente alteradas, probablemente por deshidratación leve. Se tomaron cultivos de la piel y fueron positivos para Klebsiella pneumoniae, iniciando tratamiento antimicrobiano específico. El examen dermatológico confirmó el diagnóstico de bebé colodión.
Su atención a nivel cutáneo incluyó mantener la membrana para evitar deshidratación; la paciente se bañaba diariamente con una barra neutra dermolimpiadora sin aroma, y se le aplicaban emolientes después del baño y, cada cuatro horas, una crema a base de pantenol. Fue necesario colocar lágrimas artificiales en los ojos para mantener las córneas húmedas; se dio tratamiento para el dolor vía intravenosa. Se alimentó a través de una sonda nasogástrica con fórmula hidrolizada con triglicéridos de cadena media que garantizaba el mayor aporte calórico y una mayor ingesta de proteínas.
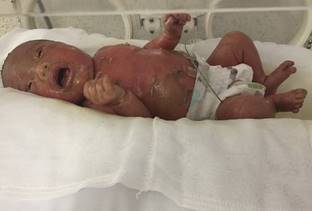
La evolución fue lenta pero favorable después de una semana. Se administró emoliente con urea al 5% y ácido glicólico para favorecer el desprendimiento de la membrana (Figura 4). La paciente fue dada de alta después de 28 días, tras lograr la estabilización de signos vitales, remisión del proceso infeccioso, y desprendimiento de la corteza. Se dio seguimiento de forma multidisciplinaria en Pediatría, Dermatología Pediátrica, Genética, Psicología, Oftalmología y Audiología.
Discusión
La ictiosis lamelar es una forma de ictiosis verdadera muy poco frecuente, con un cuadro clínico característico que permite el diagnóstico, abordaje y tratamiento precoz. En muchos de los casos no existen antecedentes familiares de ictiosis, pero con frecuencia se asocia a consanguinidad entre los padres. En el presente caso existían antecedentes de consanguinidad y una hermana mayor con un cuadro de ictiosis similar, lo que hizo considerar el componente genético de esta condición.6
La biopsia cutánea correlacionada con la prueba genética molecular no sólo es útil en el diagnóstico de la ictiosis laminar, sino también ayuda para determinar el riesgo genético de la enfermedad en la familia. Sin embargo, dado que el diagnóstico es meramente clínico, este tipo de análisis deben ponderarse de acuerdo a la situación familiar ya que pueden ser costosos.7
Los 12 genes que se sabe que están asociados con ARCI son ABCA12, ALOX12B, ALOXE3, CASP14, CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7, SLC27A4 y TGM1;8 sin embargo, alrededor del 15% de las familias afectadas no tienen variantes patógenas en alguno de estos genes. Un panel multigen que incluye estos genes es la prueba de diagnóstico de elección. Si no está disponible, se pueden considerar pruebas de un sólo gen, comenzando con ABCA12 en individuos con ictiosis arlequín, TGM1 en los que tienen ARCI sin presentación de arlequín al nacer y SLC27A4 en aquéllos con síndrome de ictiosis-prematuridad.9
El presente caso, desde nuestra perspectiva, parece corresponder a un caso familiar de ictiosis congénita, siendo muy probablemente de tipo ictiosis lamelar con un modelo de herencia autosómica recesiva. Lo anterior, basado en los hallazgos clínicos, patológicos y los antecedentes heredofamiliares.
La infección de la piel es la morbilidad asociada más frecuente como complicación de la enfermedad, siendo la sepsis su manifestación más grave.7 En el presente caso, la RN tuvo infección por Klebsiella, la cual se resolvió con tratamiento dirigido sin otras complicaciones.
A continuación, señalamos los cuidados generales al nacimiento de este grupo de pacientes:
Tratamiento de las manifestaciones: Los RN se deben colocar en un ambiente húmedo en una incubadora, tener precauciones de contacto para prevenir infecciones y dar tratamiento a las mismas; se recomienda el uso de cremas/ungüentos emolientes para mantener la piel suave, flexible e hidratada.10 Para niños mayores, se indica humidificación con baños largos, lubricación y agentes queratolíticos como alfahidroxiácidos o preparados de urea para promover la descamación y el adelgazamiento del estrato córneo;11 para aquéllos con ectropión, lubricación de la córnea. En el caso de pacientes con afectación cutánea grave, el uso de retinoides puede ser una opción.12
Prevención de complicaciones secundarias: se deben instaurar medidas preventivas a estos RN, para evitar infecciones, deshidratación, sobrecalentamiento y afectación de la córnea. La liberación de la membrana de colodión en los dedos puede ser apropiada para mantener la circulación, y a nivel del tórax puede coadyuvar a una respiración adecuada.10
Vigilancia: Examen físico regular para detectar evidencia de infección. A edades mayores, se recomienda observar lesiones de piel ya que en estos pacientes se incrementa el riesgo de carcinoma de células escamosas, carcinoma de células basales, nevos melanocíticos atípicos o melanoma maligno.11
Asesoramiento genético: ARCI se hereda de manera autosómica recesiva. Cada hermano de un individuo afectado tiene un 25% de probabilidad de serlo también, un 50% de posibilidades de ser un portador asintomático y un 25% de expectativa de no verse afectado y no ser un portador.13 En los casos que ya han sido identificadas variantes patogénicas relacionadas con ARCI en una familia, pudiera ser conveniente realizar pruebas de portador para parientes en riesgo, así como en embarazos con mayor riesgo.6

