











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
In recent years, there has been growing interest in using pressure as a specific variable to not only study but also discover various material properties, such as superconductivity, magnetic behavior, and structural phase transitions. The CeCu2Si2. compound is part of a group of materials that have received considerable attention, called the ThCr2Si2. type, which was first reported by Ban and Sikirica [1,2], known for its diversity of intriguing physical phenomena, including unconventional superconductivity, heavy fermion states, valence fluctuations, quantum criticality and multiple magnetic transitions. Before we delve into our cover-age, it is worth acknowledging a variety of outstanding reviews and thought-provoking, comprehensive papers dedicated to these materials. Initially, the heavy-fermion metal CeCu2Si2. stood as a groundbreaking discovery in unconventional superconductivity, not reliant on phonon mediation. For a considerable period, it was thought to exhibit single-band d-wave superconductivity, based on various measurements suggesting a nodal gap structure [3]. A crystallo-graphic investigation of CeCu2Si2., having the same structure as the ThCr2Si2.compound, was conducted in Ref. [4], providing atomic parameters for self-consistent Linear Muffin Tin Orbital band structure calculations for CeCu2Si2. and the isostructural LaCu2Si2.. Furthermore, AFe2As2.compounds, with A representing divalent alkaline-earth or rare-earth metals, exhibit superconductivity under specific conditions such as chemical doping or external pressure, while maintaining a tetragonal ThCr2Si2-type structure [5]. In a separate study [6], fifteen new compounds of the AB 2X2.type were synthesized, where A signifies a lanthanoid, B denotes Fe, Co, or Ni, and X represents P, As, or Sb. These compounds crystallize with the ThCr2Si2-type structure. In another investigation [7], a phase transition from a tetragonal phase to a collapsed tetragonal phase in EuCo2As2 was observed at 4.7 GPa. In the case of CeNi2Ge2, the temperature-dependent magnetic susceptibility demonstrates a Curie-Weiss-like behavior above 2 K, indicating the absence of a magnetic transition above this temperature. The effective magnetic moment, p eff , and the paramagnetic Curie temperature, θ p , are determined to be p eff = 2.18μ B and θ p = -12 K, respectively. This suggests that most of the Ce ions are in the Ce3+ state, with a partial presence of Ce4+ [8]. The overlap between the valence and conduction bands near the Fermi level confirms the metallic character of SrCo2Si2, as detailed in reference [9]. In the case of BaNi2As2, its high dielectric constant makes it a promising material for high-value capacitor production. Using data on elastic constants, the calculated Debye temperatures are found to be 323.70 K for BaNi2P2 and 272.94 K for BaNi2As2.
However, the thermal conductivity of these compounds is relatively low, at 0.56 for BaNi2P2 and 0.46 for BaNi2As2 [10]. Pugh’s ratio and Poisson’s ratio calculations indicate the ductile properties of Ca/YRh2Ge2 and the brittleness of Sr/BaRh2Ge2, while the Cauchy pressure suggests ductility across all these phases [11]. The maximum dimensionless figure-of-merit ZT for the α-BaCu2S2-type material reached 0.30 at 773 K for x = 0.03, with a power factor of 0.61 mW/mK2, which is notably high for a ThCr2Si2-type structure [12]. Transport property measurements reveal that BaMn2Sb2 exhibits a room temperature Seebeck coefficient of approximately 225 μ V /K, although the low electrical conductivity results in unfavorable thermoelectric properties. More recently, theoretical research on various materials has been published using the ab initio method to determine electronic and optical properties. On the other hand, the semi-classical Boltzmann transport theory in the BoltzTraP code was used to demonstrate the thermoelectric properties [13-20] corresponding to the experimental results. To the best of our knowledge, no theoretical research has been conducted on all aspects of the physical properties of the CeCu2Si2 compound. Therefore, in this work, physical properties such as structural, electronic, optical, thermodynamic, and thermoelectric properties will be studied to fill the gaps in the literature. Therefore, this paper is organized as follows: in Sec. 2, we describe the details of the computational method. In Sec. 3, we examine the obtained results for the structural, electronic, optical, thermodynamic, and thermoelectric properties of the studied materials. Section 4 is devoted to conclusions.
2. DFT study
The first-principles approach FPLAPW (full potential linearized augmented plane wave) implemented in the Wien2k code is used to calculate the physical characteristics of CeCu2Si2 [21]. In our computations, we used the density functional theory (DFT) Kohn-Sham approach (KS method). For exchange-correlation interactions, we employ the modified Becke-Johnson (mBJ) and local spin density approximation (LSDA) functional calculations [22]. LSDA has a flaw in its ability to predict the properties of excited states, despite being the best theory for interpreting experimental evidence. Thus, we exploited the mBJ potential to obtain a better approximation of the band gaps. In order to ensure the various physical parameters, state density, and energy gaps, the calculations were performed using a thousand k-points. The Boltzmann transport equation was integrated with the energy band structure that was produced by applying the transport distribution function theory as it was implemented in the BoltzTraP code [23]. As illustrated in Fig. 1a), CeCu2Si2 is a member of the ThCr2Si2 family of structures and has a body-centered tetragonal structure with space group I4/mmm (no. 139). Table I also has a list of the atomic locations. Figure 1b) shows the energy variation of the CeCu2Si2 versus the volume. The resulting lattice strain p was then defined as the ratio of the change in the lattice constant c to the lattice constant c 0 after the tensile force was applied in the c-direction [24]:
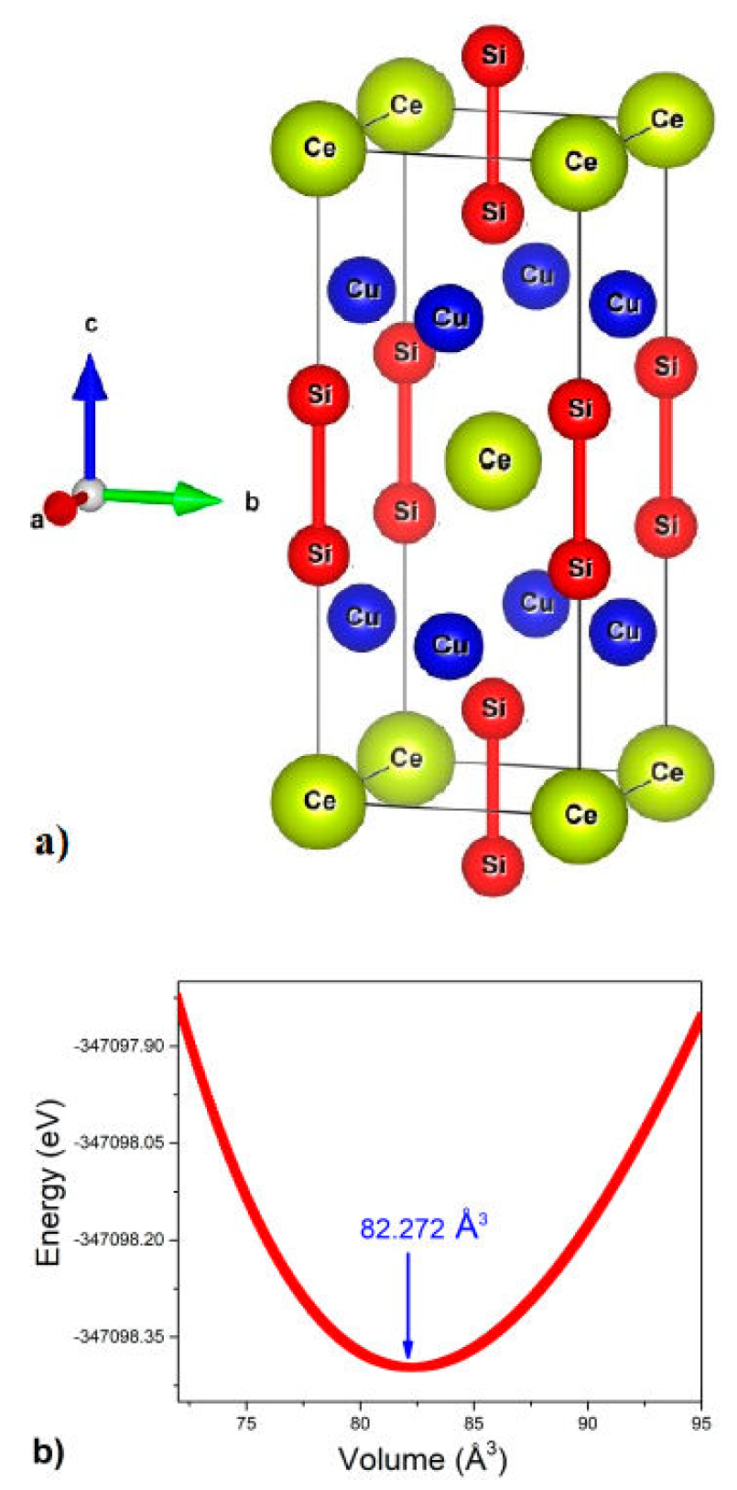
Figura 1 a) A sketch of the CeCu2Si2 structure. b) The energy variation vs the volume of the CeCu2Si2.
3. Results and discussions
3.1. Total energy and lattice parameters
Prior to examining CeCu2Si2’s electrical transport characteristics, the bulk modulus, lattice constants, and static pressure transitions are determined by structural optimization. The empirical Birch-Murnaghan equation of state is used to carry out the optimization [25]. The compound under study has its optimal properties obtained, namely: bulk modulus B, lattice constants a 0 and c 0, volume V 0, first pressure derivative B’, and minimal total energy E 0. Table II presents a summary of these parameters’ values. This table showed that the out-comes of the experiment and the current findings agreed fairly well.
3.2. Elastic properties
Elastic properties of solids relate to the mechanical and dynamical behavior under stress and create opportunities for industrial applications. The elastic properties of CeCu2Si2 material were calculated in this work using the WC-GGA method [26]. The details of the calculated elastic parameters such as elastic constants C ij , bulk B, shear G, Young’s E moduli (in GPa), and Poisson’s ratio ν are listed in Table III. The mechanical stability condition of hexagonal crystals requires the following conditions: C 11 > |C 12|, C 44 >0, (C 11 + C 12)C 33 > 2(C 13)2, and (C 11 - C 12)C 66 > 2(C 16)2 [27]. Since C 11 is smaller than C 33 for CeCu2Si2, we can infer from the elastic values obtained that the c-axis is more stiff than the a-axis. Furthermore, we observe that the unidirectional compressive strength along the major crystallographic axes is greater than the shear strain, as indicated by the C 11 and C 33 constants being larger than the C 44 and C 66 constants. Pugh’s research [28] suggested using the ratio of the bulk to shear modulus B/G to forecast the ductility behavior of materials. The material is ductile if the B/G ratio is greater than 1.75. If not, it exhibits fragile behavior. The B/G ratio of 1.62 under ambient settings indicates that CeCu2Si2 under unrestrained conditions is more brittle.
3.3. Electronic properties
Using the LSDA-mBJ potential, we further investigated the density of states of the CeCu2Si2 compound to investigate the origin of the band structure. A key component in determining the electrical characteristics of these materials is their electronic density of states (DOS). As seen in Figs. 2a) - 2e), the total density of states (TDOS) and the partial density of states (PDOS) for each of the elements Ce, Cu, and Si are computed. The involvement of each atom in the band structure is depicted in these figures, along with every possible linear combination of their atomic orbitals. States of the element Ce play a significant role in the conduction band, whereas states of the element Cu dominate the valence bands. The absence of a bandgap supports the compound’s metallic nature and is consistent with the inference made from Figs. 2e) band structure. The non-magnetic compound is further guaranteed by the symmetrical contributions at the top and bottom of the TDOS.
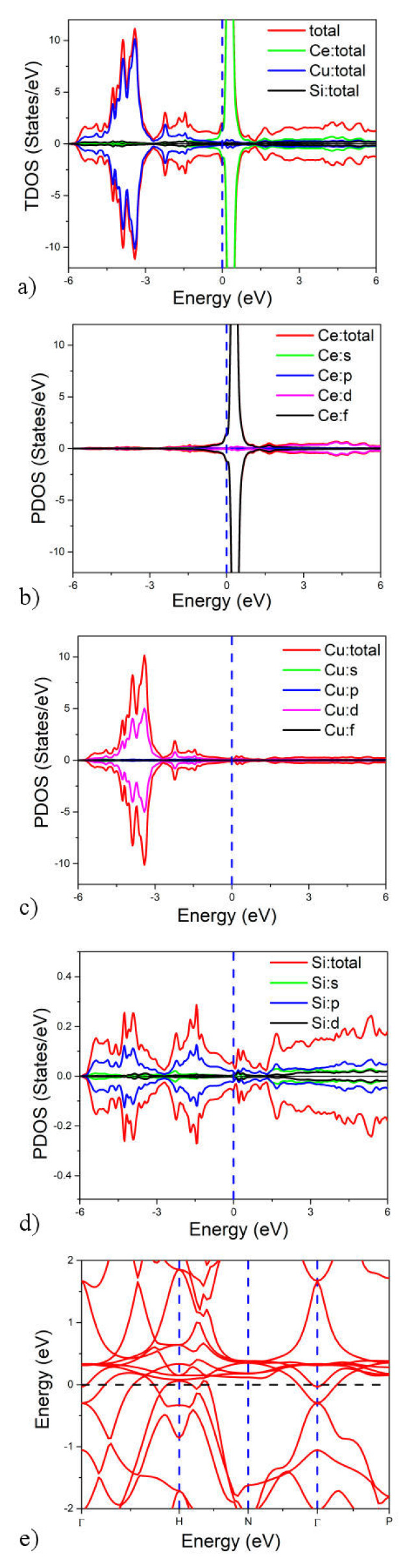
3.4. Optical properties
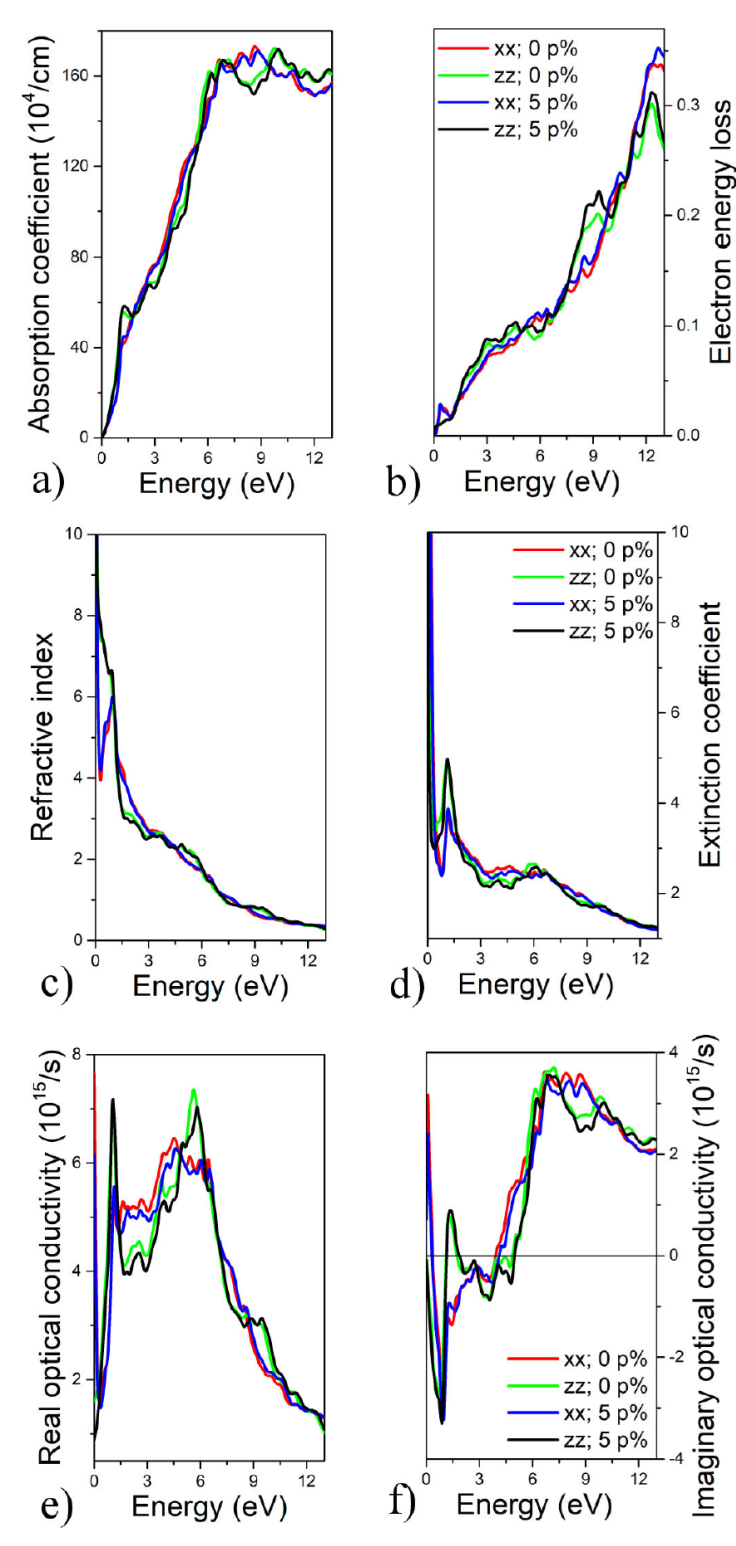
The investigation of the optical properties of CeCu2Si2 is of great significance for exploring their potential applications in photovoltaic devices. To analyze the optical characteristics, various parameters such as energy electron loss, refractive index, extinction coefficient, absorption coefficient, and the real and imaginary parts of the dielectric tensor and optical conductivity are examined. Fig. 3 presents the results obtained from the study of these optical properties using the LSDA- mBJ approach, as discussed in Refs. [29, 30]. The absorption coefficient is a crucial optical property that plays a significant role in energetic materials and solar cells. It provides valuable insights into the extent to which light of a specific wavelength, with a well-defined energy, can penetrate a material before being absorbed. This property is particularly relevant for optimizing the efficiency of solar energy conversion. As the sun emits light at various frequencies in the form of photons, understanding the absorption coefficient helps us evaluate how effectively a material can harness solar energy. The behavior of the calculated absorption coefficient of CeCu2Si2 has been depicted in Fig. 3a). It has been observed that the curves increase in intensity when photon energies are higher and show a single peak in the Infrared domain. The higher one observed is the xx-direction, and its intensity is 175 × 104 cm-1. Particularly, high absorption and quick photo-response make this compound an excellent choice for photovoltaic applications. The peak in the energy loss function reflects the dual nature of plasma resonance, which includes the associated frequency called plasma frequency. Above the plasma frequency, the material behaves as a dielectric, while below it exhibits metallic characteristics. This peak provides valuable information about the transition between the dielectric and metallic behavior of the material. The electron energy loss function for CeCu2Si2 is presented in Fig. 3b) with and without strain in the direction of xx and zz. The highest energy loss function occurs in the infrared region when 5% of strain is applied in the direction xx. It is well known that these peaks are due to inter-band transitions between various high symmetry points. The refractive index is a dimensionless quantity that quantifies the transparency of a material to incident photons. It represents the speed at which light propagates through the material, providing a measure of how much the light is bent or refracted when passing through the material. Figure 3c) shows the refractive index for the xx and zz directions under strain and without strain of CeCu2Si2. All curves decrease because of the increase in energy. The optical energy that is absorbed in the optical medium during light propagation is described by the extinction coefficient. As illustrated in Fig. 3d), extinction coefficients are presented for the xx and zz directions under strain and without strain of CeCu2Si2. All curves decrease with increasing energy. We can see that the directions xx and zz, with and without strain, give the same shape of the extinction coefficient. The property of the material known as optical conductivity determines the link between the amplitude of the inducing electric field and the current density induced at any given frequency. The real and imaginary optical conductivity parts for the xx and zz directions under strain and without the strain of CeCu2Si2 are shown in Figs. 3e), f). In the real part, all the peaks show an increase till a maximum peak where the curve of the xx direction under 5% of strain at 7.2 × 1015 s-1 is the highest one. Following that, the lines begin to flatten out as the energy level rises. All the curves of the imaginary part of optical conductivity show negative values at low energy and increase to positive values. The graph demonstrates that light absorption causes an increase in conductivity in the Infrared domain. The characteristic could be used in material photocells.
3.5. Thermoelectric properties
The BoltzTraP code combined with the semi-classical Boltzmann theory is used to determine the thermoelectric (TE) behavior of CeCu2Si2. Thermoelectric (TE) materials have the capability to generate valuable power by harnessing waste heat that would otherwise dissipate into space during regular operation. Notably, these materials are environmentally friendly, positioning them as crucial energy resources for future developments and playing a significant role in sustaining our energy needs. The Seebeck coefficient S, electrical conductivity (σ/τ), and electronic part of the thermal conductivity (κe/τ) were used to characterize the transport properties of CeCu2Si2. Employing two current approaches [23], we have estimated the whole Seebeck coefficient as expressed below:
Figure 4a) illustrates the temperature-dependent fluctuation of the Seebeck coefficient for CeCu2Si2 in both spin states, both with and without 5% strain. All curves exhibit an upward trend with rising temperature. Given the positive See-beck coefficient, indicating an n-type material, CeCu2Si2’s total Seebeck coefficient proves valuable in encapsulating the alloy’s thermoelectric characteristics comprehensively. In contrast, Fig. 4b) presents the electrical conductivity of CeCu2Si2, considering both total and spin states, represented by the relaxation time (σ / τ) against temperature, with and without strain. Notably, all curves demonstrate an increase in electrical conductivity with temperature, with 5% strain further enhancing this conductivity. These findings highlight the material’s advantageous high electrical conductivity and low resistivity, minimizing losses in electrical charge transport due to the Joule effect an essential characteristic for its viability as a thermoelectric substance. Examining the electronic part (κe / τ) of CeCu2Si2’s thermal conductivity in Fig. 5c), both spin-up and spin-down scenarios are compared at varying temperatures, considering the presence or absence of 5% strain. The electronic thermal conductivity consistently rises with temperature across all curves. Furthermore, the application of 5% strain leads to a notable increase, reaching its peak value of 2.3×1015 W·K-1 ·m-1 ·s-1 at 400 K.
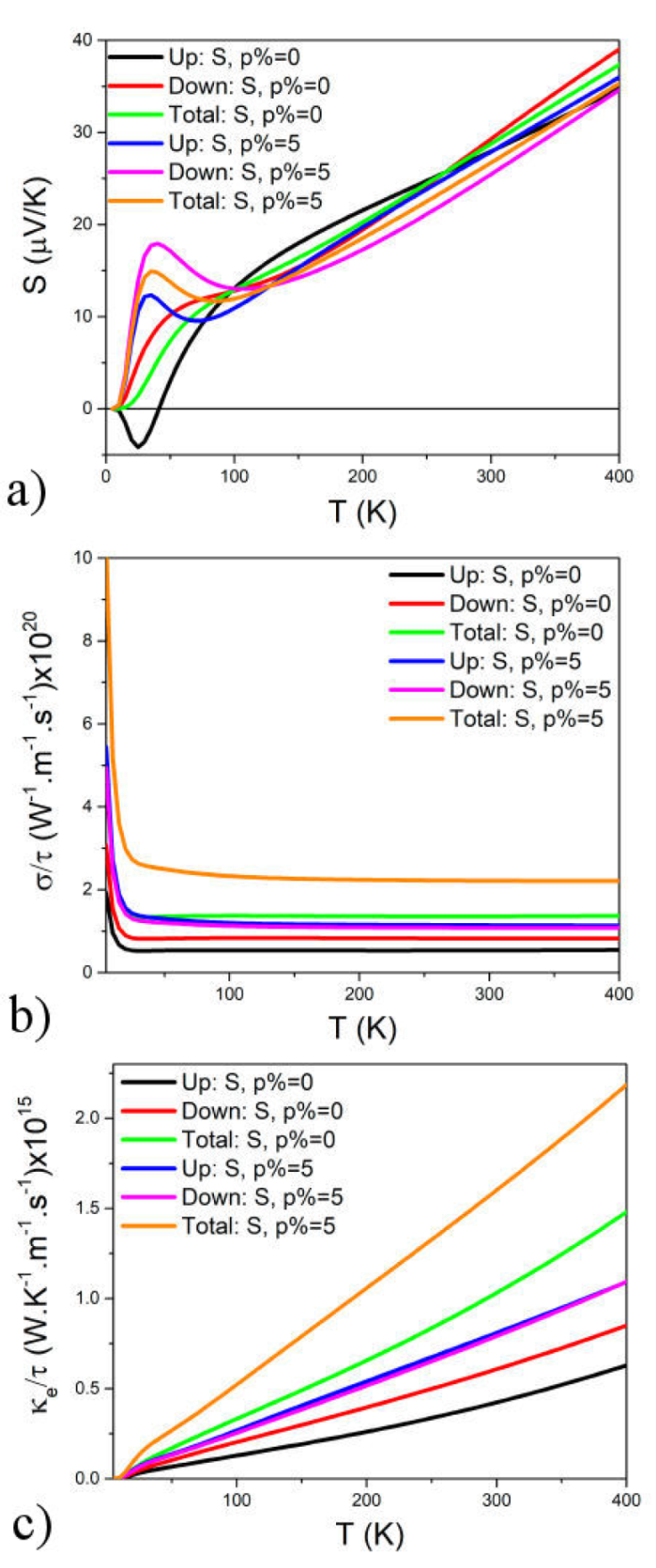
Figure 4 The Seebeck coefficient a) Electrical conductivity, b) electron thermal conductivity, and c) as a function of the temperature for CeCu2Si2.
3.6. Thermodynamic properties
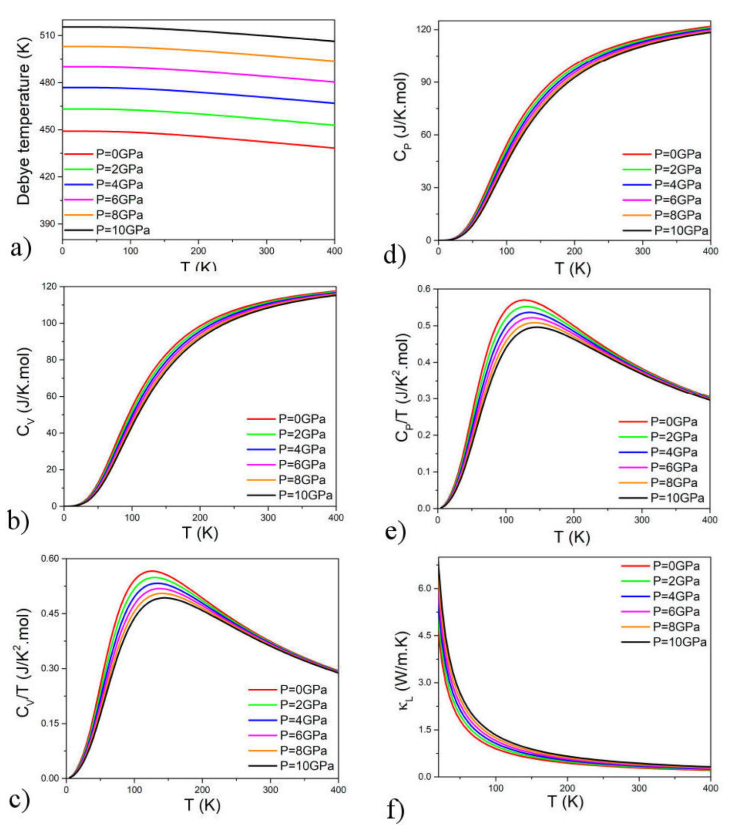
The intrinsic properties of materials, as described by thermodynamics, play a pivotal role in the realm of material science and engineering. In this specific examination, the thermal characteristics of CeCu2Si2 are scrutinized utilizing the quasi-harmonic Debye model, encompassing temperature spans from 0 to 400 K and strain variations from 0 to 10 GPa. A crucial parameter in this model is the Debye temperature (θ), which offers valuable insights into diverse physical attributes of solids, such as heat capacity, elastic factors, thermal expansion rate, and entropy. Additionally, it is intricately linked to specific heat, structural stability, lattice vibrations, and the strength of chemical bonds [31]. The Debye temperature (θ) has been computed, from the elastic constants, using the relation:
V at is the volume per atom, ρ is the density, and K and G are the bulk and shear modulus, respectively. The Debye temperature was calculated from the full elastic constant tensor averaged by the Reuss-Voigt-Hill scheme (K = 192.6 GPa, G = 99.2 GPa) using Eq. (2). In Fig. 5a), the relationship between the Debye temperature and tensile strain has been depicted. The Debye temperature increases proportionally with increasing tensile strain. Additionally, the curves of the Debye temperature exhibit a decreasing trend with increasing temperature. These findings indicate that the introduction of dilatation strain can enhance the mechanical and thermal stability of CeCu2Si2. Within the Debye model, the Grüneisen parameter could be calculated using the quasi-harmonic relation:
Figures 5b) and 5d) show the heat capacity at constant volume and the heat capacity at constant pressure for different strains. All the curves show a similar shape for different pressures and exponentially increase with temperatures. In Figs. 5c) and 5e), the heat capacity at constant volume per unit temperature and the heat capacity at constant pressure per unit temperature for different strains are plotted. All the curves show an increase at low temperatures and a decrease at higher temperatures. In addition, the lattice thermal conductivity κ L is calculated by using the Debye temperature θ D , and the Grüneisen coefficient (γ) is computed using Gibbs2 software [32]. The Method also implements an outstanding relationship to compute the part of the thermal conductivity κ L by using the Slack approach described by the following equation
where A is a set of physical parameters. Following Ref. [33], it may be calculated as follows
In the context of this discussion, θ D represents the Debye temperature, γ denotes the Grüneisen coefficient, V signifies the volume per atom, T stands for temperature, n represents the number of atoms in the primitive unit cell, and M corresponds to the atomic mass. In Fig. 5f), the thermal conductivity of the lattice κ L for CeCu2Si2 exhibits an exponential decrease with rising temperature and an increase with elevated pressure. The substantial κ L value indicates the presence of an anti-harmonic effect within the material. With a notably low lattice thermal conductivity, it can be asserted that the current material holds great promise for applications in thermoelectricity.
4. Conclusions
In conclusion, we have conducted a comprehensive review of the CeCu2Si2 compound, focusing on various physical attributes. Our study concentrated on the structural, electronic, elastic, optical, thermodynamic and thermoelectric properties of this compound. To carry out this research, we harnessed the power of density functional theory (DFT) as implemented in the Wien2k software package. We used the WC-GGA method to calculate the elastic properties of the CeCu2Si2 material. This reveals that CeCu2Si2 is unconstrained and tends to exhibit brittleness. Furthermore, the metallic nature of the compound was corroborated by the density of states analysis. This is consistent with our conclusions from examining the band structure. Additionally, we examined various optical characteristics, including electronic energy loss, absorption coefficient, refractive index, extinction coefficient, as well as real and imaginary optical conductivity. Our results revealed the remarkable absorption qualities of the compound in the infrared spectrum. Additionally, the thermoelectric results indicated that the compound exhibits p-type behavior. This is characterized by positive values of the Seebeck coefficient. These results were meticulously analyzed, providing invaluable information on the properties of the CeCu2Si2 compound.

