











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
An enhancer is a short region of DNA that can bind to specific proteins to activate gene transcription. The first enhancer was discovered in 1981 in the simian virus 40 (SV40). This enhancer was identified as a 72-base pair (bp) tandem repeat sequence in the SV40 genome, capable of increasing the ectopic expression of a reporter gene by hundreds of times1. Subsequently, enhancers were identified in mammalian mouse genes in 19832. Shortly after, various enhancers were discovered in different cells and tissues. Enhancers can positively regulate gene expression during development through cis- or trans-interactions3,4.
The activity of enhancers is often associated with specific chromatin characteristics. Active enhancers typically bind to transcription factors (TFs)5 and are marked by histone H3 lysine 4 monomethylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac), whereas lacking histone H3 lysine 4 trimethylation (H3K4me3)6. With the rapid advancement of high-throughput sequencing technologies, numerous sophisticated methods have been developed for predicting active enhancers. Among these, chromatin immunoprecipitation sequencing (ChIP-seq) is the most widely used. In addition, emerging technologies such as High-throughput chromosome conformation capture, Cleavage Under Targets and Tagmentation, and Assay for Transposase-Accessible Chromatin with high-throughput sequencing have been developed to identify the activation status of enhancers7.
The term "super-enhancers" (SEs) was first introduced in 2004 by Chen and colleagues. They identified a 651 bp DNA fragment in the baculovirus genome capable of stimulating reporter gene promoter activity by up to 7,000-fold8. It was not until a decade later, in 2013, that the concept of SEs was formally proposed by Young's team at the Whitehead Institute for Biomedical Research in the United States9. Subsequent studies have demonstrated that SEs play a critical role in maintaining cell identity and driving cancer development10. This article begins by examining the structure and characteristics of SEs, introducing methods for their identification and related databases, and exploring their potential mechanisms of action. It further discusses the acquisition of disease-specific SEs and reviews recent advancements in the study of SEs in various neurodegenerative diseases. The aim is to provide insights and guidance for future research on the role of SEs in neurodegenerative diseases.
STRUCTURAL FEATURES AND IDENTIFICATION OF SUPER-ENHANCERS
The discovery of SEs represents a deeper exploration of the enhancer concept. The term was first refined in the research of Professor Richard A. Young. Through studies on mouse embryonic stem cells (ESCs), Young observed that interactions between several enhancers and their bound TFs could sustain the pluripotency of ESCs. These enhancer clusters were found to activate exponentially the expression of their target genes, which are often critical for determining cell identity. Consequently, Whyte et al9. defined these clusters of enhancers with potent transcriptional activation properties as SEs. Compared to typical enhancers, SEs exhibit higher levels of H3K27ac and H3K4me1 modifications, greater sensitivity to TF binding, and denser association with transcriptional activators Mediator Complex Subunit 1 (MED1) and Bromodomain-containing protein 4 (BRD4). These components interact to form transcriptional complexes, collectively driving robust gene expression11,12 (Fig. 1). Furthermore, the constituent enhancers within a SE can function independently13, whereas interactions between these components are critical for their collective activity14. The deletion of a core enhancer within a SE can result in the inactivation of the entire SE, ultimately leading to the suppression of its target gene15.
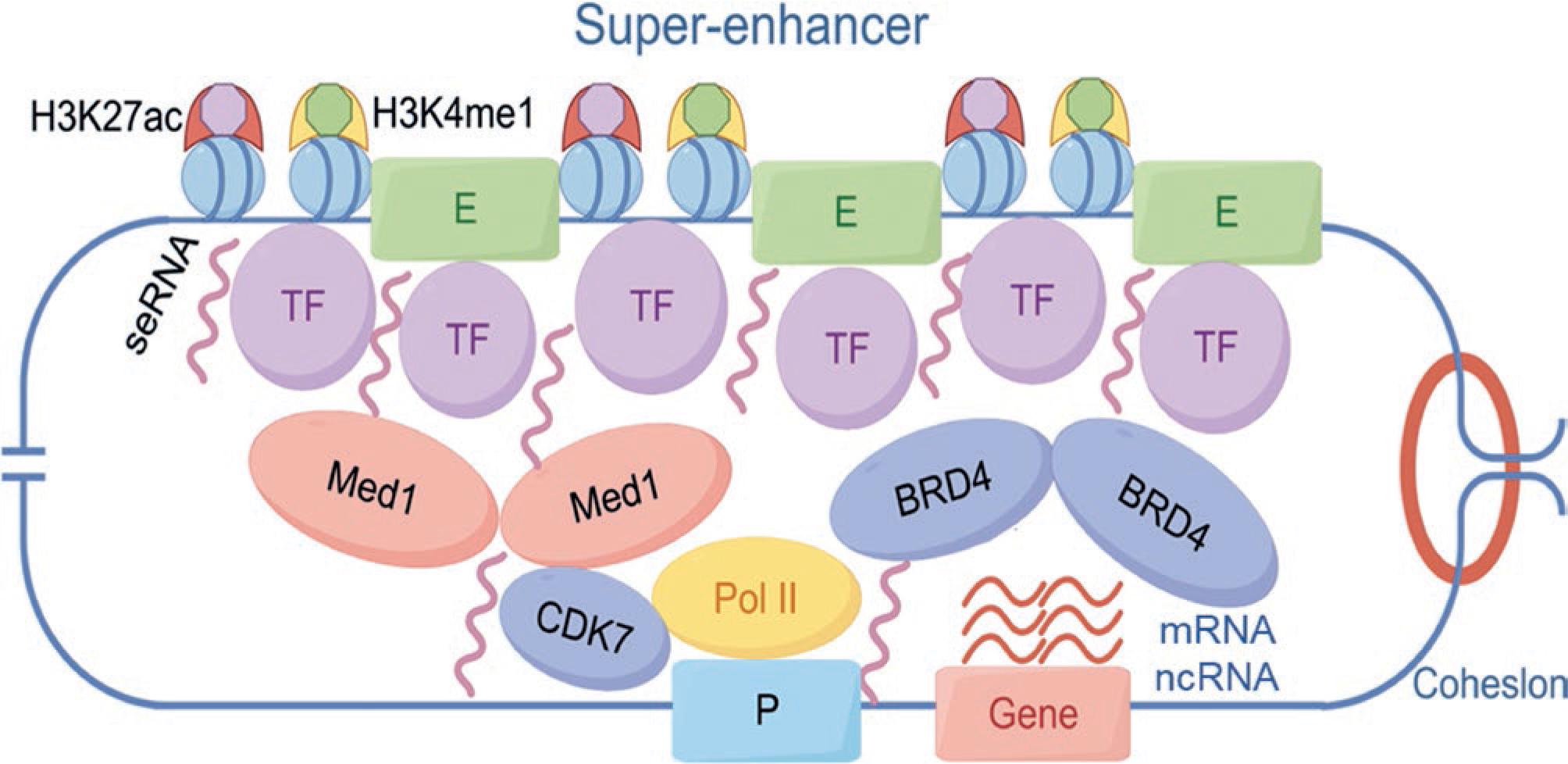
Figure 1. Structure and function of SEs. SEs orchestrate robust gene expression through the coalescence of transcription factors, coactivators (MED1 and BRD4), chromatin markers (H3K27ac and H3K4me1), and super-enhancer RNAs (seRNAs). These core SE components, in association with CDK7, Pol II, the cognate promoter, and the target gene locus, undergo liquid-liquid phase separation to establish a transcriptionally active biomolecular condensate. Within this spatially confined compartment, cooperative interactions among transcriptional machinery and chromatin elements synergistically amplify the efficiency of RNA polymerase II-mediated gene expression.
At present, the identification of SEs primarily relies on the levels of histone modifications on enhancers and the binding intensity of transcriptional coactivators. ChIP-seq is commonly used to analyze and identify SEs by profiling histone modifications such as H3K27ac and H3K4me1, as well as the binding of the mediator complex16. The Rank Ordering of SEs (ROSE) algorithm, developed by Young's laboratory, is one of the most widely used techniques for identifying SEs. The process consists of five key steps13: (1) Perform H3K27ac ChIP-seq: Conduct ChIP-seq experiments for H3K27ac in the cell type or tissue of interest. (2) Map ChIP-seq Data: Align the H3K27ac ChIP-seq data to the reference genome. (3) Identify Active Enhancers: Determine regions of active enhancers based on ChIP-seq peaks. (4) Merge Enhancers within 12.5 kb: Use the ROSE software to merge enhancers located within a 12.5 kb range. (5) Rank Enhancers by ChIP-seq Signal: Rank the merged enhancers and remaining single enhancers based on their ChIP-seq signal intensity. This process identifies the threshold separating SEs from typical enhancers17,18. It is important to note that when merging enhancers within the 12.5 kb range, regions within ± 2.5 kb of transcription start sites should be excluded to prevent the inclusion of promoter regions in the analysis (Fig. 2).
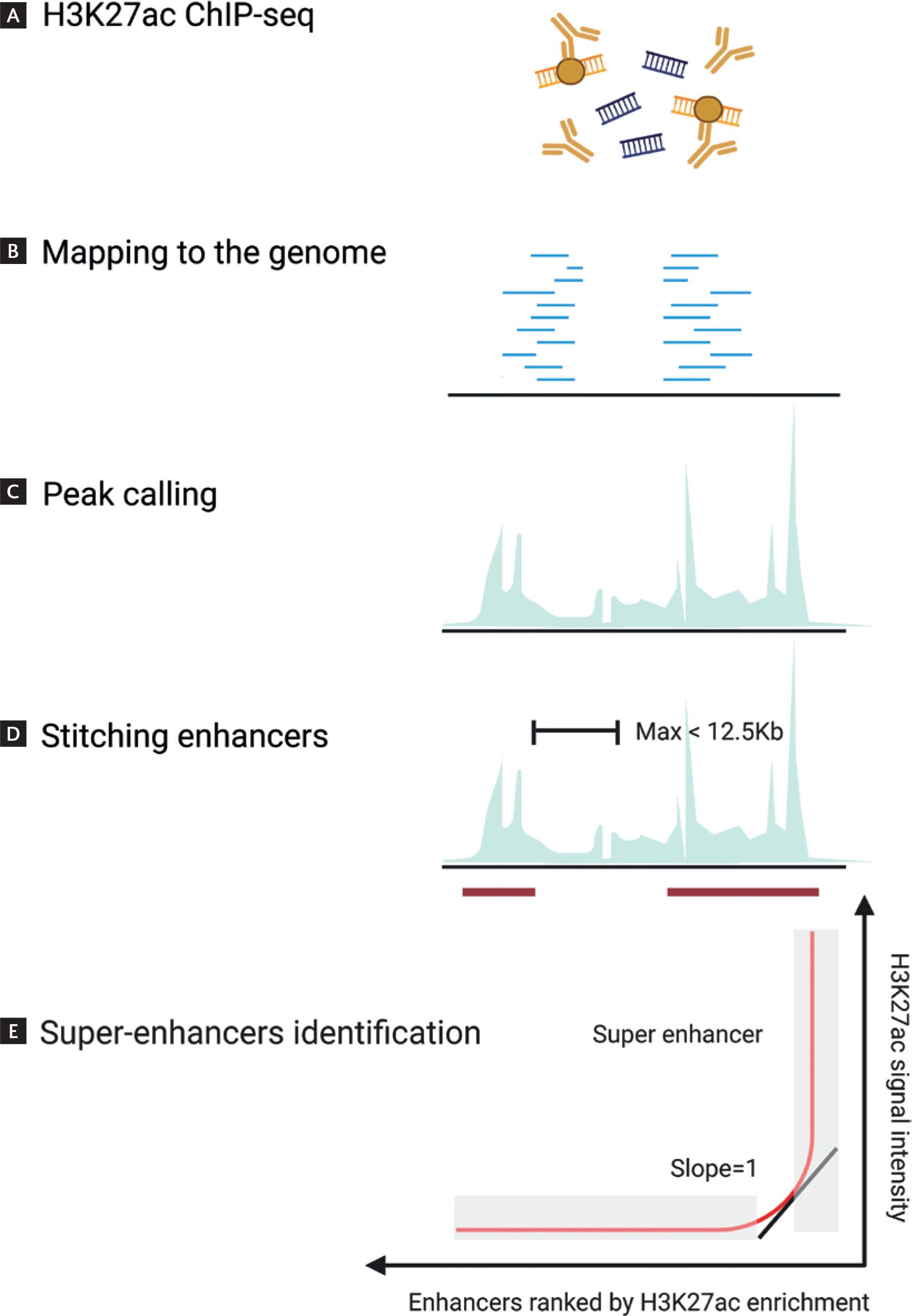
Figure 2. SEs identification workflow. A: the canonical SEs identification pipeline begins with H3K27ac ChIP-seq profiling in the target cell type or tissue to capture chromatin regions marked by active enhancer signatures. B and C: sequential reads are aligned to the reference genome, followed by peak calling to identify candidate enhancers. D: neighboring enhancer elements within a 12.5 kb distance are stitched into continuous domains using the ROSE algorithm. E: the merged enhancer clusters are then ranked by their H3K27ac signal intensity, enabling differentiation of super-enhancers (characterized by exceptionally high signals) from typical enhancers. This integrative bioinformatics approach prioritizes large transcriptional regulatory hubs critical for cell identity maintenance and disease pathogenesis.
DATABASES RELATED TO SUPER-ENHANCERS
In recent years, several databases related to SEs have been developed, providing researchers with extensive SE information. Among these, the dbSUPER database (a database of SEs in mouse and human genome) (http://asntech.org/dbsuper/) is the first integrated and interactive SE database, established in 2016. This database incorporates SEs from two primary sources: 1. Published SEs: SEs reported in scientific publications and supported by experimental data. 2. Identified SEs: SEs were identified through the analysis of publicly available H3K27ac ChIP-seq data using the ROSE algorithm. To date, dbSUPER has cataloged 82,234 SEs derived from 102 human cell types/tissues and 25 mouse cell types/tissues19. This resource serves as a valuable tool for advancing the understanding of SEs and their roles in gene regulation and disease. The SEA (SE Archive) database (http://sea.edbc.org) was first established in 2016 and has since undergone multiple updates, incorporating SEs data from a wide range of species. At present, in version 3.0, the database includes a total of 164,545 SEs identified across 11 different species and 266 cell types/tissues/diseases. Notably, the latest version features 80,549 newly identified SEs, 52 of which have been experimentally validated. As one of the most comprehensive SE databases available, SEA provides an invaluable resource for studying SEs across diverse biological contexts20,21. In 2019, Professor Chunquan Li's research team from the First Affiliated Hospital of the University of South China developed SEdb 1.0 (A comprehensive SE database of human and mouse), a database that annotated the functional impact of SEs on gene transcription regulation in a cell type-specific manner. SEdb 1.0 included extensive human SE data and detailed annotations. Recently, the team released the updated SEdb 2.0 (http://www.licpathway.net/sedb), which supports data for over 2,500 tissues/cell types from 2,670 samples, encompassing more than 1.7 million SEs (human: 1,167,518; mouse: 550,226). This includes data from 541 primary human samples, 1,198 newly published human samples, and 931 mouse samples. SEdb 2.0 integrates a wide array of cutting-edge genetic and epigenetic features, such as chromatin states and 3D chromatin interactions, offering the most comprehensive SE annotation to date22. This robust database serves as an invaluable resource for advancing research into SE functions and regulatory mechanisms.
MECHANISMS AND FUNCTIONAL ROLES OF SUPER-ENHANCERS
As a specialized form of enhancer, the fundamental regulatory mechanism of SEs involves enhancer-promoter interactions. These interactions form a three-dimensional chromatin structure that regulates gene expression through the coordinated actions of TFs, cofactors, and enhancer-promoter looping23. Super enhancers, comprising clusters of constituent enhancers, rely on the collective activity of these individual components to exert their function. Each constituent enhancer independently binds to TFs and cofactors, and the cumulative interactions with promoters ultimately drive the transcriptional regulation of target genes24.
Super enhancers can regulate the expression of target genes through non-coding RNAs (ncRNAs). These functional RNA transcripts are not translated into proteins and play critical roles in various biological processes, including cell differentiation, tumorigenesis, apoptosis, and metabolism25. Studies have shown that SEs indirectly regulate the expression of protein-coding genes by modulating the transcription of non-coding RNAs (ncRNAs). Among ncRNAs, microRNAs (miRNAs) were the first to be identified. Research has demonstrated that SEs can activate miRNA transcription by facilitating the processing of precursor miRNAs by the DROSHA enzyme. These miRNAs, in turn, regulate the expression of their target genes26. For example, brown adipose tissue contains specific SEs that regulate the expression of miR-32. Elevated miR-32 expression suppresses the transcription of its target gene TOB1, which subsequently activates the p38/MAPK signaling pathway. This activation promotes the thermogenic activation of brown adipose tissue and the browning of subcutaneous white adipose tissue, enhancing the body's thermogenic response. These processes help mitigate obesity and metabolic syndrome27. In addition to miRNAs, long non-coding RNAs (lncRNAs) can also be regulated by SEs. Studies have shown that lncRNAs can form RNA:DNA:DNA triplex structures between SEs and promoters, thereby recruiting TFs to the SE region and facilitating its activation28. These findings highlight that the regulation of non-coding RNAs is a crucial aspect of the functional mechanism of SEs.
Studies have revealed that SEs can transcribe enhancer RNAs (eRNAs), but the expression levels of these eRNAs are significantly higher compared to those produced by typical enhancers. These high-expression eRNAs are referred to as SE RNAs (seRNAs)29. seRNAs can regulate the expression of adjacent genes through cis-acting mechanisms. For example, a distal SE located 45 kb upstream of the NANOG gene has been shown to regulate both NANOG and its neighboring gene DPPA3 in Murine ESCs. Researchers found that the seRNA produced by this SE specifically regulates DPPA3 expression by stabilizing the interaction between the SE and the DPPA3 locus30. In addition to regulating neighboring target genes through cis-acting mechanisms, seRNAs can also induce target gene expression through trans-regulatory effects. For instance, MyoD upstream non-coding RNA (MUNC) associated with the MYOD gene has been identified as an enhancer RNA. MUNC has been shown to induce the production of specific myogenic transcripts, demonstrating its role in trans-regulatory gene activation. However, in cell lines where MUNC was knocked out, it was observed that overexpression of MUNC could still induce the production of various myogenic transcripts. This suggests that MUNC can function independently of MYOD despite being classified as an eRNA of MYOD. Furthermore, these findings indicate that MUNC also acts as a trans-regulatory lncRNA, extending its regulatory role beyond its association with MYOD31. In summary, the majority of seRNA mechanisms are similar to those of eRNA; however, our understanding of seRNA remains limited. Further research is needed to elucidate its roles and regulatory mechanisms in greater detail.
In addition to the regulatory mechanisms involving non-coding RNAs and seRNAs, the concept of phase separation has emerged as a prominent focus in exploring the mechanisms of SE function. Phase separation was initially described as a physical phenomenon and was first applied to explain biological functions in 2009. This was based on research in Caenorhabditis elegans, where a type of protein known as P granules were discovered to exist not in a conventional solid state but as dynamic, liquid-like condensates. P granules exhibited liquid-like properties, including fusion, dripping, and wetting behaviors. This experiment was the first to demonstrate that phase separation could serve as a fundamental physicochemical mechanism underlying the organization of the cytoplasm32. In 2017, Hnisz et al. applied the theory of phase separation to explain the regulatory mechanisms of SEs. This marked a significant advancement in understanding how SEs function in gene regulation. The team demonstrated that the SE-specific transcriptional coactivators BRD4 and MED1 form nuclear puncta at SE sites, exhibiting liquid-like properties. Through phase separation, BRD4 and MED1 create a compartment that sequesters transcriptional components, thereby stabilizing the transcription process33. A growing body of experimental evidence highlights the phase separation of SEs as critical to their functional activity. SE phase separation drives efficient gene expression through the dynamic assembly of transcriptional condensates, a mechanism broadly exploited in cancer to sustain malignant phenotypes. For example, in head and neck squamous cell carcinoma, the cytoskeleton regulator RNA (CYTOR), a long intergenic non-coding RNA, facilitates FOSL1-dependent SE phase separation to promote metastasis34. In lung adenocarcinoma, SP1 undergoes phase separation to activate SEs, accelerating cancer progression35. Disrupting these condensates effectively suppresses oncogene expression. Small-molecule inhibitors of BRD4 or MED1 can destabilize phase separation, inhibit SE activity, and downregulate oncogenes33,36. While molecular details and therapeutic applications require deeper exploration, targeting phase separation has emerged as a novel strategy in cancer treatment.
In summary, SEs participate in diverse physiological and pathological processes through distinct mechanistic pathways. While substantial experimental evidence over recent years has elucidated their varied molecular mechanisms, precise characterization remains challenging due to ambiguities in their definition and molecular composition. With the identification of SEs and advances in their study, researchers have increasingly focused on unraveling their structural architecture, functional dynamics, and mechanistic underpinnings37. Consequently, SEs have emerged as a focal point of investigation in stem cell biology and oncology.
MECHANISMS FOR THE ACQUISITION OF DISEASE-SPECIFIC SUPER-ENHANCERS
The acquisition of disease-specific SEs in disease-related cell types can be explained by three models. The first model involves the activation of SEs due to genetic mutations. For instance, studies have shown that in T-cell acute lymphoblastic leukemia (T-ALL), the activation of a single TAL1 allele results in the formation of an abnormal 20 kb SE. This acquired SE contains binding sites for leukemogenic TFs such as RUNX1, GATA-3, and TAL1 itself. This alteration is caused by a 12 kb insertion, which creates new transcriptional activator binding sites38. In the second model, genetic mutations do not occur at the TF binding sites but rather at the binding sites of the CCCTC-binding factor (CTCF), which defines the boundaries of SEs. The loss of CTCF binding disrupts these boundaries, leading to the activation of nearby silenced genes and potentially resulting in disease. Another study by Hnisz et al. found that in T-ALL, base deletions result in the loss of boundary CTCF binding, leading to the activation of previously silenced oncogenes39. The third model proposes that during somatic hypermutation, the activation of activation-induced cytidine deaminase (AID) contributes to genomic instability, which can lead to the development of malignancies. A comprehensive analysis of the molecular mechanisms indicates that AID can trigger genomic translocation, relocating an oncogene adjacent to a SE. This positions the oncogene under the control of the SE, resulting in its overexpression and the development of B-cell lymphoma40.
ROLE OF SUPER-ENHANCERS IN NEURODEGENERATIVE DISEASES
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), are a group of age-related progressive disorders characterized by the selective, disease-dependent loss of specific neuronal regions and/or subpopulations41. Before neuronal loss occurs, there is a prolonged period of neuronal dysfunction, during which significant changes also take place in glial cells, including the development of neuroinflammatory responses42. These changes are associated with epigenetic and transcriptional dysregulation, characterized by alterations specific to certain cell types43. Notably, an increasing body of research supports the idea that alterations in SE regulation play a critical role in the transcriptional dysregulation observed in PD, HD, and AD.
ROLE OF SUPER-ENHANCERS IN ALZHEIMER'S DISEASE
AD is the most common progressive neurodegenerative disorder, characterized by the accumulation of amyloid-beta and neurofibrillary tangles, which are composed of abnormally phosphorylated Tau protein. AD leads to progressive synaptic and neuronal loss, particularly in the prefrontal cortex and hippocampus, resulting in memory decline44. The risk of developing AD is 60-80% genetically determined, with over 40 genetic risk loci identified to date45. For example, variants in the genes encoding amyloid precursor protein (APP) and apolipoprotein E4 (APOE4) are among the most significant risk variants associated with the disease46. Genome-wide association studies (GWAS) have identified additional genetic risk variants, including those associated with APOE and BIN147,48. Analysis of GWAS databases indicates that nearly 30% of non-coding single nucleotide polymorphisms (SNPs) associated with AD are located within enhancers. Furthermore, 95% of AD-associated SNPs are found in enhancers that colocalize with expression quantitative trait loci genes within the same topologically associated domains49. Nott et al. conducted an epigenomic analysis using cortical tissue from AD patients and found that AD risk variants are enriched in microglia-specific enhancers50. Thus, accumulating evidence suggests that brain-specific enhancers, particularly microglia-specific enhancers, contribute to the pathogenesis of AD in a cell-type-specific manner48-51. Studies have identified 27 SNP mutations associated with AD pathology, five of which are located within SE regions52. This finding suggests that SEs may play a significant role in AD, although the specific molecular mechanisms remain to be elucidated.
ROLE OF SUPER-ENHANCERS IN PARKINSON'S DISEASE
PD is the second most common neurodegenerative disease worldwide. Its primary pathological hallmark is the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc), accompanied by dopamine depletion53. Research indicates that ferroptosis, a form of iron-dependent cell death, is involved in the degeneration of dopaminergic neurons in PD54. In another study, SEs were found to be involved in the ferroptosis pathway of PD. Using ChIP-seq and the ROSE algorithm, the authors identified specific SEs in the SNc of a 6-OHDA-induced PD rat model. Among these, the most prominent was the SE regulating the Sorting Nexin 5 (SNX5) gene. Subsequent experiments demonstrated that silencing the identified SE significantly reduced SNX5 expression and decreased ferroptosis levels in 6-OHDA-induced PD cell models. These findings suggest a link between disease-specific SEs and the pathogenesis of PD55. Another study revealed that during the expression of the SNCA gene, which encodes α-synuclein (the main component of Lewy bodies), an enhancer cluster resembling a "super-enhancer" forms within an intron of the SNCA gene. Deletion of this enhancer cluster led to a reduction in SNCA expression, suggesting that targeted editing of disease-specific SEs could serve as a potential therapeutic strategy for PD56.
ROLE OF SUPER-ENHANCERS IN HUNTINGTON'S DISEASE
HD is a neurodegenerative disorder characterized by the loss of striatal neurons caused by an expansion of CAG repeats (> 35 CAG) in the first exon of the huntingtin (HTT) gene. This leads to progressive impairments in motor control, cognition, and emotional regulation57. Epigenomic studies using brain tissues from HD mouse models and HD patients have revealed significant findings. Notably, widespread changes in H3K27ac levels were observed in the striatum of HD R6/1 transgenic mice58,59. H3K27ac ChIP-seq analysis of postmortem striatal tissues from early-stage HD patients revealed similar findings. The study showed that, compared to controls, H3K27ac levels were reduced near SEs in HD neurons. This reduction was associated with the downregulation of genes regulated by these SEs in neurons58,59. In contrast, H3K27ac levels were upregulated at glial cell-specific SEs, leading to increased transcription of genes regulated by these SEs60. In summary, H3K27ac data from HD suggest that HD mutations induce the loss of activity in neuron-specific SEs that regulate neuronal identity genes while simultaneously increasing the activity of glial cell-specific SEs. These findings highlight the critical role of cell-specific SEs in the regulation of HD pathogenesis and progression.
CONCLUSION AND FUTURE PERSPECTIVES
With the aging population on the rise, neurodegenerative diseases have become a significant threat to the health of elderly individuals, particularly in China. However, due to the limited understanding of their underlying pathogenesis, effective therapeutic strategies remain elusive. With advancements in technology and deeper research, evidence suggests that specific SEs may play a critical role in the progression of neurodegenerative diseases and represent a potential class of therapeutic targets. Super enhancers are defined as a class of cis-regulatory elements with exceptionally strong transcriptional activation capabilities. Compared to typical enhancers, they possess more robust regulatory functions, driving the expression of genes that define cell identity. While extensive research on SEs has been conducted, there remains a lack of clear and universally accepted criteria for defining them. At present, SEs are primarily distinguished from typical enhancers by calculating differences in molecular signal intensity markers on active enhancers13. However, further experimental validation is still required17. In recent years, significant progress has been made in understanding the role of SEs in cancer. Various SE inhibitors, such as cyclin-dependent kinase inhibitors and bromodomain and extra terminal protein inhibitors, have been developed to interfere with tumor progression and suppress metastasis61,62. However, research on SEs in neurodegenerative diseases remains relatively limited. By leveraging established SE database platforms19-22 and integrating neurodegenerative disease-related epigenomic data for joint analysis, researchers can gain deeper insights into the functions and regulatory mechanisms of disease-specific SEs. This approach lays a solid foundation for further exploration of the roles of SEs in neurodegenerative diseases.

