











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
The adoption of next-generation sequencing (NGS) in medicine has greatly enhanced diagnostic rates of genetic diseases over the past decade1-3. NGS allows to refine the diagnosis and improve patient management by permitting the rapid screening of hundreds or thousands of genes simultaneously at a lower cost compared with sequential testing of multiple genes. Since about 85% of currently known disease-causing mutations are located within exonic regions4, whole-exome sequencing (WES), an NGS technique that analyzes protein-coding regions of all human genes, is being increasingly employed in clinical settings for precise diagnosis of genetic disorders5-7. Particularly, WES has become a powerful and affordable technique to improve the molecular diagnosis of monogenic diseases8-10. In addition, the concurrent development of bioinformatics software for appropriate and accurate data analysis and classification of genetic variants according to their expected biological pathogenicity has greatly increased the rates of definitive molecular diagnosis in these disorders11-13. Unfortunately, genome sequencing technologies, which are becoming indispensable in clinical settings, are not widely available in the developing countries14.
Genetic eye disorders, affecting around one in 1000 people worldwide15, encompass a clinically and genetically heterogeneous group of diseases that collectively involve every structure of the eye, ocular adnexa, and visual system16. Genetic eye disorders can occur at all ages and are frequently associated with severe visual deficiency. As per the World Health Organization, genetic eye disorders are one of the top 10 major causes of global ocular health burden17. It is estimated that up to 30% of the approximately 6000 Mendelian diseases in humans have some ocular involvement and thus there are hundreds of genes causing eye diseases when mutated18. Many inherited eye diseases have considerable genetic heterogeneity (i.e., multiple genetic causes), and therefore, the use of NGS allows a growing number of patients to obtain a molecular diagnosis for their disease19-22.
Here, we evaluated the diagnostic yield of WES in a group of 90 unrelated Mexican individuals with suspected genetic eye disorders. In addition, we assessed the improvement of diagnostic rates by reanalysis of WES data in patients without an initial molecular diagnosis, at least 1 year after the first data analysis.
METHODS
Patient enrollment
A total of 90 patients with ocular disorders of probable monogenic origin were included in the study. Ocular disorders of monogenic origin were suspected due to: more than 1 person in the same family affected with an ocular disease; having a suspected diagnosis related to a monogenic etiology; an individual with a clinical diagnosis of a syndrome with a genetic origin; and a history of parental consanguinity. Subjects were recruited at the Department of Genetics of the Institute of Ophthalmology “Conde de Valenciana,” Mexico City, during the period from January 2019 to December 2020. The participant selection criteria were as follows. Inclusion criteria: individuals with a suspected monogenic disease, of any gender and age, with or without a family history of the disease. Exclusion criteria: individuals with multiple malformations without previous karyotype or microarray study, individuals with a recent history (< 60 days) of blood transfusion, and individuals with suspected monogenic disease due to somatic mosaicism. All participants signed an informed consent form and underwent standardized medical history records and physical examinations. Parental consent was obtained for patients under 18 years of age. The study was approved by the Institutional Ethics Committee. All patients were Mexican Mestizo, defined as a person who was born in Mexico, has a Spanish-derived last name, and has a family of Mexican ancestors going back to the third generation.
WES analysis
Genomic DNA was extracted from a 3-5 mL sample of venous blood. Genomic DNA isolation was performed with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. The obtained DNA was resuspended in 200 µL of water, quantified with a NanoDrop equipment, and stored in the DNA bank at −20°C for later use. Whole-exome sequencing of samples was performed as an external service at the 3Billion company (Seoul, South Korea). Briefly, exonic regions of all human genes (~22,000 genes) were captured with the xGen Exome Research Panel v2 kit (IDT, Coralville, Iowa, USA). The libraries were quantified by fluorimetry and were sequenced in a NovaSeq 6000 equipment (Illumina, San Diego, CA, USA) at a minimum depth of ×50. The analysis of the raw sequencing data, including alignment to the reference genome GRCh37/hg19 and the calling and annotation of variants, was performed with free access software and with the EVIDENCE program (3Billion Inc.).
Once the sequencing files were received, we identified and classified variants of clinical importance using the Franklin platform (https://franklin.genoox.com/clinical-db/home); this annotation database contains several human population databases as gnomAD (http://gnomad.broadinstitute.org/), the 1000 Genomes Project (http://browser.1000genomes.org), dbSNP (http://www.ncbi.nlm.nih.gov/snp), as well as in silico prediction algorithms as SIFT (http://sift.jcvi.org), FATHMM (http://fathmm.biocompute.org.uk), and MutationAssessor (http://mutationassessor.org).
Variant prioritization
The clinical phenotype, evidence of pathogenicity, and mode of inheritance were considered during the process of variant filtration. The variants were classified into five categories: “pathogenic (P),” “likely pathogenic (LP),” “uncertain significance (VUS),” “likely benign (LB),” and “benign (B)” in accordance with the American College of Medical Genetics and Genomics (ACMG) guidelines. The variants of clinical importance identified, classified as pathogenic or likely pathogenic, and that have not been previously reported in the literature, were investigated in a set of 400 alleles from Mexican subjects analyzed by in-house exomes, to exclude the possibility that they were benign variants typical of our population. The possible pathogenicity of missense variants or variants located within 10 nucleotides adjacent to the exon/intron junctions, which were not found included in public databases and, which were also not identified in the panel of 400 control alleles, were analyzed in silico with various computer programs. Causative variants were confirmed by Sanger sequencing following standard methods. Segregation of the candidate variant(s) in DNA of available affected relatives was achieved by Sanger sequencing. In a subset of nine cases, additional diagnostic procedures, including gene panel sequencing and copy number variant (CNV) analysis, were performed.
RESULTS
Sample characteristics
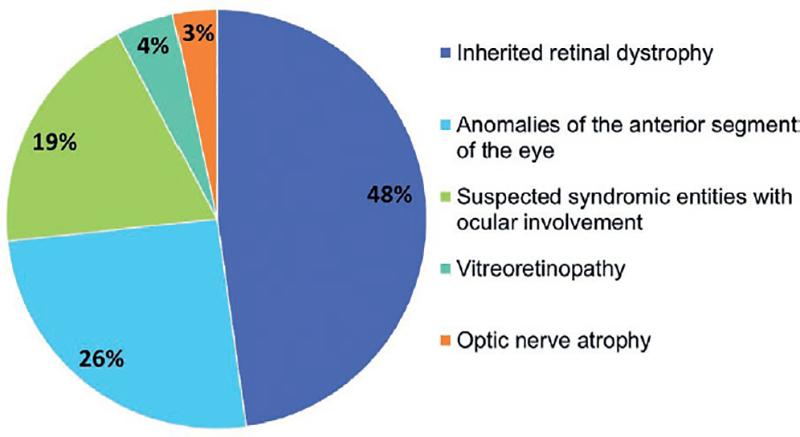
A total of 90 unrelated individuals of Mexican-Mestizo descent participated in the study; 47 (52%) were male and 43 (48%) were female. The age of the participants ranged from 1 to 60 years, and the mean age at recruitment was 20 years. A total of 61 patients (68%) were sporadic cases while 29 (32%) had a history of first-degree relative(s) with the same disease. All individuals had clinically diagnosed disorders with several degrees of ocular involvement (Fig. 1): 43 (48%) cases had hereditary retinal dystrophies, including 21 (23%) individuals with isolated retinitis pigmentosa, 13 (14%) with macular dystrophy, 3 (4%) with syndromic retinitis pigmentosa, 3 (4%) with cone-rod dystrophy, 2 (2%) with cone dystrophy, and 1 (1%) with choroideremia. There were also 23 (26%) subjects with anomalies of the anterior segment of the eye, including 7 (8%) with microphthalmia/anophthalmia, 4 (5%) with bilateral congenital cataract, 3 (4%) with bilateral congenital glaucoma, 2 (3%) with lens subluxation, 2 (3%) with corneal dystrophies, and one case each of the following diagnoses: microspherophakia, sclerocornea, and Axenfeld-Rieger syndrome (3%). In addition, there were 17 (19%) cases with suspected syndromic disorders with variable ocular involvement, 4 (4%) cases of vitreoretinopathies, and 3 (3%) with optic nerve atrophy/hypoplasia.
Diagnostic yield of WES
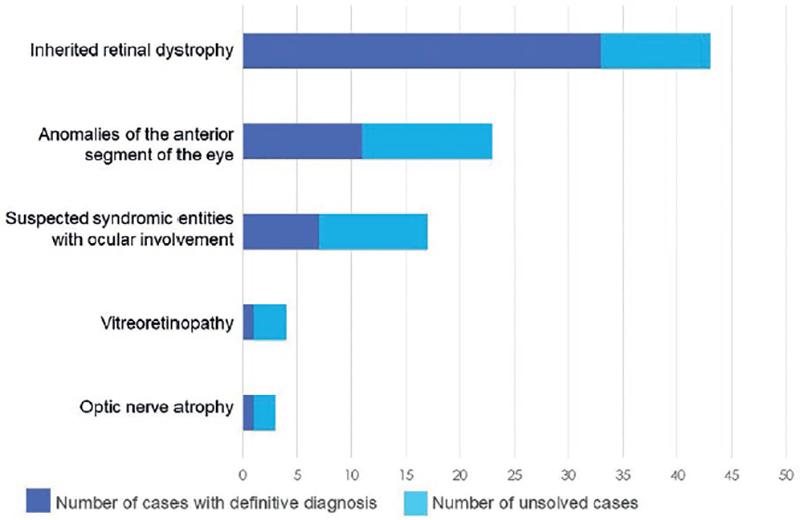
A definitive molecular diagnosis was reached in 58% (52/90) of the probands after WES data analysis (Fig. 2). The diagnostic rates among disease categories were: 77% (33 out of 43 cases) for hereditary retinal dystrophies; 48% (11/23) for anomalies of the anterior segment of the eye; 47% (8/17) for syndromic disorders with ocular involvement, 33% (1/3) for optic nerve atrophy/hypoplasia, and 25% (1/4) for vitreoretinopathy.
At the initial bioinformatics analysis, a pathogenic or likely pathogenic causal variant was identified in 46 (51%) individuals, who received the following definitive diagnoses: macular dystrophy in 12 (13%), retinitis pigmentosa in 10 (11%), anterior segment dysgenesis in 3 (3%), Usher syndrome in 2 (2%), isolated lens subluxation in 2 (2%), cone-rod dystrophy in 2 (2%), and one case of each of the following diagnoses: Leber congenital amaurosis, cone dystrophy, choroideremia, congenital stationary night blindness, Stickler syndrome, primary congenital glaucoma, syndromic microphthalmia, optic atrophy, congenital cataract, Waardenburg syndrome, Alport syndrome, Charcot-Marie-Tooth disease, juvenile hyaline fibromatosis, Lhermitte-Duclos disease, and Bloom syndrome.
In other six cases, the causal variant was identified after reanalysis of the WES data 12 months after the primary analysis, increasing the percentage of solved cases by 7%. The final diagnoses in the individuals solved in the reanalysis included: Leber congenital amaurosis (1 case), oculocutaneous albinism (1), CHARGE syndrome (1), Noonan syndrome (1), Schuurs-Hoeijmakers syndrome (1), and adrenoleukodystrophy (1).
In 4 out of 9 cases that remained unsolved after reanalysis, gene panel sequencing and CNV analysis allowed the recognition of the causal genetic defect: a deletion of exons 2 to 17 of the RPGRIP1 gene was identified in a case with Leber congenital amaurosis; an Alu element insertion in the RP1 gene was recognized in one patient with retinitis pigmentosa; a non-coding pathogenic variant in the FTL gene was recognized in one case of congenital cataract; and a pathogenic point mutation in the CHM gene was identified in one case with a clinical diagnosis of choroideremia. Thus, a final diagnostic yield of 62% (56/90) was obtained in this cohort by combining WES and gene panel sequencing/CNV analysis.
In 10 patients, the clinical diagnoses were changed based on their genetic findings: two cases diagnosed as macular dystrophy were correctly assigned as early-onset retinal dystrophy and cone-rod dystrophy type 12, respectively; one patient with Leber congenital amaurosis was correctly diagnosed as oculocutaneous albinism type 2; one case with a clinical diagnosis of cone-rod dystrophy received a definitive diagnosis of retinitis pigmentosa type 12; a patient with microphthalmia, anophthalmia, and coloboma spectrum was diagnosed with CHARGE (Coloboma, Heart anomaly, choanal Atresia, Retardation, and Genital and Ear anomalies) syndrome; a patient with a presumptive connective tissue disorder was definitively reassigned as Lhermitte-Duclos syndrome; one case with a diagnosis of Nance-Horan syndrome was correctly diagnosed as Noonan syndrome type 4; a case identified as Gillespie syndrome was definitively assigned as alacrima-achalasia-intellectual disability syndrome; one case of presumptive Axenfeld-Rieger syndrome received a conclusive diagnosis of Schuurs-Hoeijmakers syndrome; and a patient with congenital pulverulent cataract received a final diagnosis of hyperferritinemia cataract syndrome.
Mutational spectrum
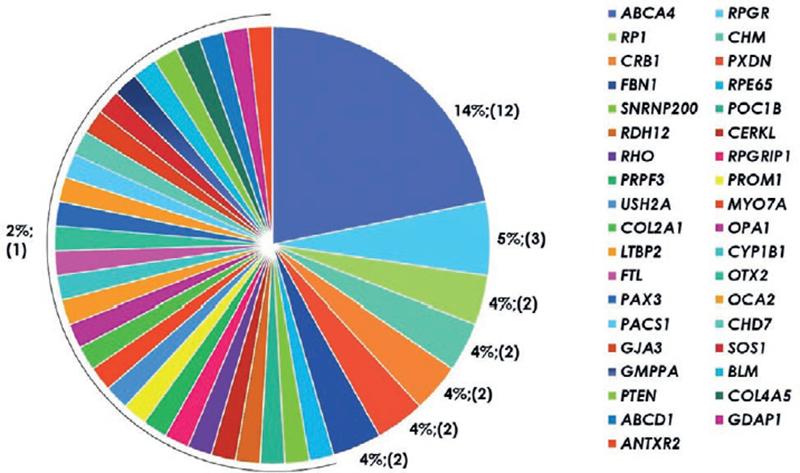
Causal variants were recognized in 38 different genes (Fig. 3, Table TS2), with a total of 71 variants identified, including 21 novel disease-causing mutations (Table TS1). According to the ACMG guidelines, 44 variants were classified as pathogenic variants and 27 were likely pathogenic variants. The identified causal mutations included 39 (55%) missense, 14 (20%) frameshift, 11 (16%) nonsense, 4 (6%) splicing site mutations, 1 (1%) synonymous variant affecting splicing, 1 (1%) non-coding, and 1 (1%) intragenic deletion. In 31 cases (56%), causal mutations affected genes associated with autosomal recessive inheritance; in 17 (31%), affected genes related with autosomal dominant inheritance; and in 7 (13%), mutations were located in X-linked genes.
DISCUSSION
Genetic testing is a process aimed to identify whether an individual has a genetic disorder or may develop one in the future. The benefits of genetic testing include the definitive diagnosis of an inherited disease, the possibility of starting a specific treatment, or initiating prevention strategies, as well as making life decisions such as career choice and family planning. In addition, genetic confirmation of a diagnosis allows for a more precise prognosis of the possible clinical evolution. Genetic testing by NGS analysis can lead to a diagnosis in days, potentially helping patients and families to avoid months or years of inconclusive and expensive medical tests23. In the present study, we assessed the effectiveness of WES for genetic diagnosis in a cohort of 90 Mexican subjects with various ocular disorders of suspected genetic origin. Inherited eye disorders are an important cause of visual impairment in children and working-age people in the Western world. Since inherited eye disorders have extensive clinical and genetic heterogeneity, genetic diagnosis based on candidate gene(s) screening is extremely difficult. WES allows examination of the coding sequences of all known human genes, enabling an efficient molecular diagnosis in patients with heterogeneous genetic diseases. Our study achieved an overall diagnostic yield of 62% (56 out of 90 cases), with 46 cases reaching a definitive diagnosis at first WES data analysis, six additional cases diagnosed after reanalysis of sequencing data 12 months after first analysis, and four other cases diagnosed by a combination of gene panel sequencing/CNV analysis. The diagnostic value of WES in genetic eye diseases has been demonstrated in previous studies. For example, Haer-Wigman et al.24 identified a genetic cause in 52% of 266 Dutch subjects with diseases causing visual impairment; Prokudin et al.25 recognized causal mutations in 36% of cases in a series of patients with developmental eye diseases; and Haug et al.26 obtained a diagnostic rate of 46% in a cohort of patients with eye malformations. The diagnostic rate obtained through WES in genetic ocular diseases depends on several factors, including the specific eye disorder, the inclusion of familial versus sporadic cases, and the occurrence of recurrent mutations in a specific population (i.e., founder mutation effect). An important aspect to consider is that the number of WES-based studies in inherited retinal dystrophies is much higher than that in other ocular diseases27, with diagnostic rates ranging from 50% to 65% in inherited retinopathies screened by WES28-30. In our study, patients with inherited retinal dystrophies reached a diagnostic rate of 77% (33 out of 43 cases).
The reanalysis of exome data in this cohort increased the diagnostic yield in ~ 7%, since six additional patients were definitively diagnosed. In three of them, variant reclassification led to a final genetic diagnosis; in two other cases, reevaluation of the phenotype allowed to refine a phenotype-genotype correlation; and in one case, familial segregation of a candidate VUS led to its reclassification as a likely pathogenic one. Recent studies have shown that 10-40% of undiagnosed patients could obtain an accurate diagnosis by reanalysis of the WES data31,32. The diagnostic rate can be increased by rescreening against newly discovered disease-associated genes and variants, using family-based screening to identify de novo variants, or by reevaluation of the patient’s phenotype33. As for the cases which remain unsolved, a negative result could mean that the condition is not genetic in origin (i.e., a “true negative” result) or could also be a false negative because the genetic cause is located in genomic regions not well detected due to limitations in current WES technologies, as occurs for non-coding variants, repeat expansions, or copy-number variants34. In fact, four additional cases from our group were solved by gene panel sequencing that gave readings of greater coverage and depth, allowing the recognition of causal mutations that were missed by WES.
Many patients with genetic diseases may receive an incorrect diagnosis35,36 or live many years without a specific diagnosis37. Not having an accurate diagnosis has a negative impact on the quality of life of patients with genetic diseases, who may undergo numerous unnecessary examinations that are displeasing and costly38. In our study, 10 patients had a change in diagnosis based on their genetic findings, which was of great importance since their clinical management was improved.
Our study demonstrates that WES is an effective approach for genetic diagnosis of monogenic ocular diseases. Our results also expand the molecular spectrum linked to human genetic eye diseases by the recognition of 21 novel disease-causing variants, which are of relevance for future molecular analyses in this field. Having a molecular diagnosis is important for patients, as it ends their diagnosis odyssey, gives them the opportunity to an individualized management, and provides the information of the disease recurrence risk for future generations.

