











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Recognized as one of the most common malignant tumors in gynecology, cervical cancer (CC) attracts extensive attention for resulting in remarkable morbidity and mortality worldwide1,2. In accordance with correlative cancer statistics, the number of CC-associated deaths in developing countries is conspicuously higher than that in developed countries, with 529,800 new CC cases and 311,365 CC-related deaths in 2018, globally3,4. Although there has been advanced progress in the therapy against CC, long-term prognosis of CC patients remains unsatisfactory due to frequent recurrence, metastasis, and resistance to radiotherapy or chemotherapy5. In recent years, molecular targeted therapies have prodigiously improved the therapeutic effect in many cancers, such as breast, lung, and colorectal cancer6-8. Therefore, further probing into therapeutic targets and prognosis biomarkers is beneficial for a higher survival rate of CC patients.
Long non-coding RNAs (lncRNAs), a class of newly identified ncRNAs with a length of over 200 nucleotides, are short of protein-coding capacity because they lack an open reading structure of a necessary length9. Numerous studies have linked lncRNAs to the molecular mechanisms underlying the progression of tumors and highlighted their significance as promising biomarkers and therapeutic targets10. Accumulating evidence has suggested that lncRNAs are closely associated with almost all aspects of cancer-related biological processes, such as gene expression, cellular growth, and transcription11,12. In addition, lncRNAs are well-known for modulating the progression of carcinomas via RNA-binding proteins13. Recently, the role of lncRNAs as competing endogenous RNAs (ceRNAs) in tumor initiation and development has been stressed. lncRNAs could serve as ceRNAs to upregulate the expression of messenger RNAs (mRNAs) through decoying miRNAs14. For instance, lncRNA PVT1 knockdown inhibits the migratory and invasive abilities of esophageal carcinoma cells through the miRNA-145/FSCN1 axis15. LINC00460 promotes the malignant phenotypes of papillary thyroid cancer cells by binding to miR-485-5p and modulating Raf116. Emerging evidence shows that plenty of lncRNAs have been implicated in the development of CC such as lncRNA OIP5-AS117, SPINT1-AS118, LncCCLM19, and LINC0065720. LncRNA NR2F1-AS1 has been considered as a critical regulator in several cancers. Through the activation of the IGF-1/IGF-1R/ERK signaling, NR2F1-AS1 facilitates the angiogenesis of breast cancer cells21. NR2F1-AS1 enhances the malignant phenotypes of thyroid cancer cells through sponging miR-338-3p and upregulating CCND1 expression22, Nonetheless, the biological role and potential regulatory mechanism of NR2F1-AS1 in CC have not been clarified yet.
We hypothesized that NR2F1-AS1 may function as a ceRNA to suppress the malignant behaviors of CC cells. Hence, this study was designed to verify the suppressive effects of NR2F1-AS1 on CC cellular development and probed into the potential mechanisms related to NR2F1-AS1. This study may provide novel implications of NR2F1-AS1 as a potential biomarker for CC treatment.
METHODS
Tissue sample collection
Twenty-five paired CC tissues and matched adjacent normal tissues were collected from 18 patients with CC admitted to Binhai County People’s Hospital (Jiangsu, China). None of the patients underwent pre-operative radiotherapy, chemotherapy, or other antitumor regimens. In addition, patients who had previously been diagnosed as other types of cancers were excluded from this study. The excised tissues were quickly immersed in liquid nitrogen and stored at −80°C for further analysis. All patients gave written informed consent before the study. The experiments were approved by the Ethics Committee of Binhai County People’s Hospital (Jiangsu, China).
Cell lines
Three CC cell lines (SiHa, ME-180, and SW756) and a normal human cervix epithelial cell line (Ect1/E6E7) were commercially obtained from Chinese Academy of Sciences. Cells were cultivated in Dulbecco’s modification of eagle’s medium (DMEM) (Invitrogen, USA) including 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin in a moist incubator containing 5% CO2 at 37ºC.
Cell transfection
The NR2F1 knockdown plasmids (sh-NR2F1 #1 and sh-NR2F1 #2) and their negative control (sh-NC); the miR-642a-3p overexpression plasmids (miR-642a-3p mimics) and their negative control (NC mimics); and the NR2F1-AS1 overexpression plasmids (pcDNA3.1-NR2F1-AS1) and their negative control (pcDNA3.1-NC) were all designed by and ordered from GenePharma (Shanghai, China). Cell transfection was conducted utilizing Lipofectamine 2000 (Invitrogen, USA) as per the manufacturer’s instructions.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay
MTT assay was performed to examine cell viability as per the protocol of suppliers. Briefly, transfected cells were detached by 0.25% trypsin when cell density reached 80%. After being measured accurately, the cells (5 × 103 cells/well) were plated in 96-well plates and the culture medium containing 15 μl of MTT solution (5 mg/mL, Sigma, USA) was added into each well at the time point of 24, 48, and 72 h. After incubation for 4 h at 37ºC, the formazan crystal was dissolved in 150 μL of dimethyl sulfoxide. A Microtiter plate reader (BioTek, USA) was adapted to measure the optical density (OD) at 490 nm. A cell-viability curve graph was created with the time point as the abscissa and the OD as the ordinate.
Colony formation assay
Transfected SiHa and SW756 cells were inoculated into 6-well plates and incubated in DMEM with 10% FBS for 2 weeks. Phosphate buffered saline (PBS) was used to rinse the cells twice. The colonies formed by SiHa and SW756 cells were fixed with 4% formaldehyde for 40 min and dyed with 0.1% crystal violet for 20 min. The number of colonies defined as more than 50 cells per colony was counted under a light microscope.
Western blot analysis
RIPA lysis buffer (Thermo Fisher Scientific, China) was employed to extract total proteins, which were then quantified by a bicinchoninic acid detecting kit (Beyotime Biotechnology, Nanjing, China). About 10% SDS-PAGE was utilized for the separation of equal amount of protein samples. After been transferred onto a polyvinylidene fluoride membrane, protein samples were sealed with 5% skimmed milk for 1 h. Membranes were cultured with the primary antibodies against E-cadherin (Abcam, ab40772), N-cadherin (Abcam, ab76057), Vimentin (Abcam, ab92547), NRIF1 (Abcam, ab175932), and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam, ab8245) at 4ºC overnight. After being washed with TBST and further cultivated with secondary antibodies (Abcam, UK) at 37ºC for 1 h, the protein band was measured using the enhanced chemiluminescence reagent. Quantification of protein expression was achieved using ImageJ software23.
RNA extraction and real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Trizol reagent (Invitrogen, USA) was utilized to extract total RNAs from CC and normal cells and tissues according to the manufacturer’s instructions. The High-Capacity complementary DNA (cDNA) Reverse Transcription Kit (Thermo Fisher Scientific) was utilized to synthesize the first cDNA strand. A SYBR Green PCR Kit (Takara, Japan) was employed to conduct RT-qPCR on an ABI7500 real-time qPCR system (7500, ABI Company, USA). MiR-642a-3p expression level was normalized to that of U6. GAPDH functioned as the internal reference for NR2F1-AS1 and NR2F1. Detailed conditions were 10 min at 95ºC, and 40 cycles of 10 s at 95ºC and 30 s at 60ºC (for NR2F1-AS1, E-cadherin, Vimentin, N-cadherin, NR2F1, and GAPDH), 10 min at 95ºC, and 40 cycles of 10 s at 95ºC and 2 at 60ºC and 1 min at 95ºC, 30 s at 55ºC, and 30 s at 95ºC (for miR-642a-3p). Relative quantification of genes was calculated using the 2-ΔΔCt method24. Primers are listed below: NR2F1-AS1 forward: 5’-CCACTTCAAGGAAACACTGAC-3.’ reverse: 5’-GTCCTAGAGGCAGTGTATCTC-3’; miR-642a-3p forward: 5’- GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGGTTCC-3’, reverse: 5’-CGCGAGACACATTTGGAGAG-3’; miR-423-5p forward: 5’-GCCTGAGGGGCAGAGAGC-3’, reverse: 5’-CCACGTGTCGTGGAGTC-3’; 5’- miR-642b-3p forward: 5’-AGACACAUUUGGAGAGGGACCC-3’, reverse: 5’-UUUGAAAGAGAACCCGUGUGAG-3’; NR2F1 forward: 5’-AAACTCAGCAGACACAGAC-3’, reverse: 5’-AGAAGAGCTGCTCGATGAC-3’; GAPDH forward, 5’-CCTCCTGTTCGACAGTCAG-3’, reverse: 5’-CCCATACGACTGCAAAGAC-3’; U6 forward: 5’-CACTTTGGCAGCACATATACC-3’, reverse: 5’-CTCATTCAGAGGCCATGCT-3’.
Transwell assay
Transwell assay was implemented for evaluating cell migratory and invasive abilities. Transwell chambers (Corning, USA) with (for invasion assay) or without (for migration assay) matrigel (BD Biosciences, USA) were utilized. Approximately 2 × 104 cells were inoculated into the top compartment with serum-free medium, and the medium containing 10% FBS (600 μl) was added into the bottom compartment. After 24 h of cultivation, 4% paraformaldehyde was used for fixation of the migrated or invaded cells that were subsequently stained with 0.5% crystal violet. Assessment of cell invasive and migratory capacities was processed by counting invaded and migrated cells under an inverted microscope (Olympus).
Luciferase reporter assay
The full-length sequence or mutated fragments of NR2F1-AS1 containing the predicted miR-642a-3p binding sequence were inserted into pmirGLO luciferase vector (Promega, USA) to form NR2F1-AS1 wild type (Wt) or NR2F1-AS1 mutant (Mut). 3’UTR of NR2F1 Wt or Mut fragments bracketing the predicted miR-642a-3p binding site were cloned into pmirGLO luciferase vector (Promega, USA). Lipofectamine 2000 (Invitrogen, USA) was adopted to co-transfect miR-642a-3p mimics or NC mimics and the recombinant vectors. The relative luciferase activity was detected utilizing the Dual-Luciferase Reporter Assay System (Promega, USA).
Subcellular fractionation assay
After washing in precooled PBS, 1 × 107 SiHa and SW756 cells were harvested to confirm the subcellular localization of NR2F1-AS1. The Cytoplasmic and Nuclear RNA Purification Kit (Norgen, Belmont, USA) was employed to extract and purify the nuclear and cytoplasmic RNAs as instructed by the manufacturer25. NR2F1-AS1 content in both cell cytoplasm and nucleus were examined by RT-qPCR analysis. GAPDH served as the cytoplasmic localization control gene, and U6 served as the nuclear localization control gene.
RNA Immunoprecipitation (RIP)
RIP assay was processed using EZ-Magna RIP Kit (Millipore, USA), and RIP lysis buffer was employed to lyse SiHa and SW756 cells at 4ºC. Cell lysates were incubated with RIP buffer containing magnetic beads conjugated with anti-Ago2 antibody or negative control normal mouse immunoglobulin G (IgG; Millipore). RIP wash buffer was utilized to rinse the beads and proteinase K buffer was adopted for incubation of the complexes to remove proteins. The purified immunoprecipitated RNA was subjected to RT-qPCR for analyzing the enrichment of NR2F1-AS1, miR-642a-3p and NR2F1.
Statistical analysis
All statistical analyses were performed using the SPSS (version 20.0, SPSS Inc., Chicago, USA). Student’s t test and one-way ANOVA were applied for comparison between two groups and among multiple groups, respectively. All variables are shown as the mean ± standard deviation. Spearman’s correlation analysis was used for statistical correlation. Each experiment possesses three replications. p < 0.05 denotes statistically significance.
RESULTS
NR2F1-AS1 expression is significantly decreased in CC tissues and cells
To explore the role of NR2F1-AS1 in CC, the expression pattern of NR2F1-AS1 in CC tumor tissues was presented by GEPIA database (http://gepia.cancer-pku.cn/index.html). The findings showed that NR2F1-AS1 expression was at a low level in CC tissues (n = 306) compared with non-tumor tissues (n = 13) (Fig. 1A). The results of RT-qPCR revealed that NR2F1-AS1 expression was downregulated in CC cells (SiHa, ME-180 and SW756) relative to normal cervix epithelial cell line (Ect/E6E7) (Fig. 1B). Herein, we selected SiHa and SW756 cell lines for the following experiments on account of the lowest expression level of NR2F1-AS1 in them. In addition, the RT-qPCR analysis of NR2F1-AS1 expression in clinical CC and normal tissue samples showed that NR2F1-AS1 was downregulated in CC tissue samples versus in non-tumor tissues (Fig. 1C).
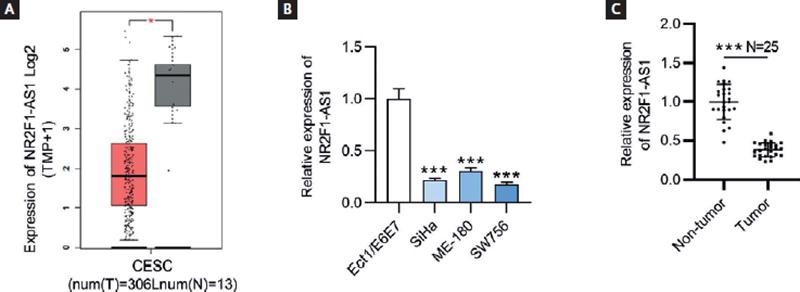
Figure 1. NR2F1-AS1 is downregulated in CC tissues and cell lines. (A) Expression profile of NR2F1-AS1 in CESC from GEPIA database. T, tumor; N, normal; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma. (B) Expression levels of NR2F1-AS1 in CC cells (SiHa, ME-180 and SW756) and normal cervix epithelial cell line (Ect/E6E7) were examined by RT-qPCR. (C) Expression levels of NR2F1-AS1 in CC and non-tumor tissues from CC patients were detected by RT-qPCR. *p < 0.05, ***p < 0.001.
NR2F1-AS1 upregulation inhibits the malignant properties of CC cells
The expression level of NR2F1-AS1 was notably upregulated in SiHa and SW756 cell lines after pcDNA3.1-NR2F1-AS1 transfection, as shown by RT-qPCR (Fig. 2A). Subsequent functional assays were employed to investigate the biological role of NR2F1-AS1 in CC cells. MTT assay demonstrated that NR2F1-AS1 upregulation significantly reduced the viability of SiHa and SW756 cells (Fig. 2B). Colony formation assay depicted that the number of colonies was significantly reduced after overexpressing NR2F1-AS1 (Fig. 2C). Consistently, pcDNA3.1-NR2F1-AS1 transfected SiHa and SW756 cells displayed markedly attenuated migratory and invasive abilities (Fig. 2D and E). Furthermore, Western blot analysis and RT-qPCR were adopted to detect the expression levels of epithelial-mesenchymal transition (EMT) process-related markers (E-cadherin, Vimentin, and N-cadherin). The results demonstrated that NR2F1-AS1 overexpression generated a notable rise of E-cadherin expression at both protein and mRNA levels as well as a dramatic decline of Vimentin and N-cadherin expression at both protein and mRNA levels in CC cells, implying that NR2F1-AS1 hindered EMT process in CC cells (Fig. 2F and G). Therefore, NR2F1-AS1 upregulation contributed to suppress CC cell malignant behaviors.
Figure 2. Overexpression of NR2F1-AS1 inhibits the malignant phenotypes of CC cells. (A) The transfection efficacy of pcDNA3.1/NR2F1-AS1 in SiHa and SW756 cells was confirmed by RT-qPCR. (B) Cell viability was detected by MTT assay on NR2F1-AS1 overexpression. (C) The influence of NR2F1-AS1 upregulation on cell proliferation was tested by colony formation assay. (D-E) Transwell assays were utilized for the examination of CC cell migration and invasion after transfecting pcDNA3.1/NR2F1-AS1. (F-G) Western blot and RT-qPCR were performed to evaluate the expression levels of EMT process-related markers at protein and mRNA levels after overexpressing NR2F1-AS1. **p < 0.01, ***p < 0.001.
NR2F1-AS1 positively modulates NR2F1 expression in CC
Based on the findings of the above-mentioned assays, we discovered that NR2F1-AS1 exerted tumor suppressive function in CC. As the cognate sense transcript of NR2F1-AS1, NR2F1 greatly caught our attention due to its role as an established biomarker of tumor dormancy26. The expression level of NR2F1 was firstly explored using GEPIA database, and we found the lowly expressed NR2F1 in CC tissue samples (n = 306) relative to non-cancerous tissue samples (n = 13) (Fig. 3A). RT-qPCR and Western blot analysis both indicated that compared with normal Ect1/E6E7 cells, the expression levels of NR2F1 were decreased in CC cell lines (Fig. 3B). In addition, the results of Spearman’s correlation analysis verified the positive association of NR2F1-AS1 expression with NR2F1 expression in CC (Fig. 4A). Thereafter, we wondered whether NR2F1-AS1 had a regulatory effect on NR2F1 expression in CC cells. Through Western blot analysis, the enhanced protein expression of NR2F1 was observed in NR2F1-AS1-upregulated SiHa and SW756 cells (Fig. 4B).
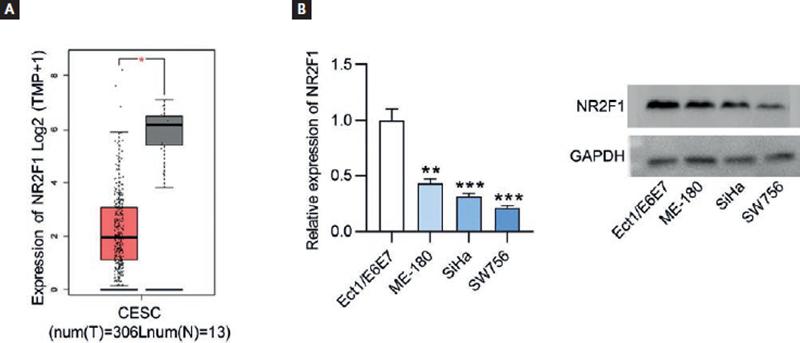
Figure 3. NR2F1 displays low expression in CC tissues and cells. (A) GEPIA database presented NR2F1 expression profile in CESC. T, tumor; N, normal; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma. (B) NR2F1 expression levels were examined by RT-qPCR and Western blot in CC cells and normal cervix epithelial cells (Ect1/E6E7). *p < 0.05, **p < 0.01, ***p < 0.001.
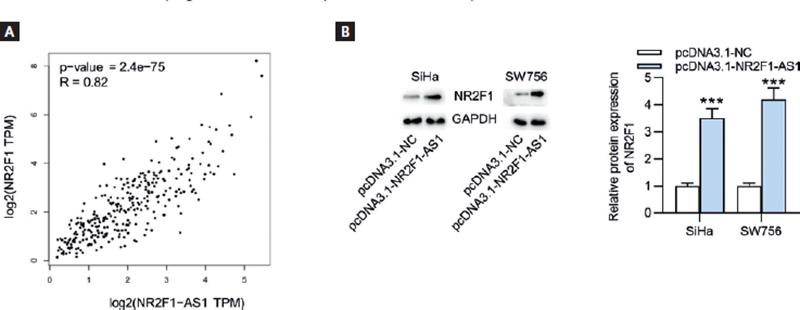
Figure 4. NR2F1-AS1 expression is positively correlated with NR2F1 expression. (A) The expression correlation of NR2F1-AS1 with NR2F1 in CC tissues was examined by Spearman’s correlation analysis. (B) Western blot analysis was used to detect the influence of NR2F1-AS1 upregulation on NR2F1 expression in CC cells. ***p < 0.001.
MiR-642a-3p is upregulated in CC cells
Afterwards, we detected the subcellular location of NR2F1-AS 1through searching LncLocator database (http://www.csbio.sjtu.edu.cn/cgi-bin/lncLocator.py), the results of which showed that NR2F1-AS1 was mainly distributed in the cytoplasm of CC cells (Fig. 5A). Subcellular fractionation assay further verified this result that more percentage of NR2F1-AS1 was found in the cytoplasm of SiHa and WS756 cells (Fig. 5B), suggesting that NR2F1-AS1 may serve as a ceRNA to sponge the corresponding miRNA and regulate the target gene expression. To further confirm the relationship between NR2F1-AS1 and NR2F1, the Ago2-RIP assay was conducted. The results demonstrated that both NR2F1-AS1 and NR2F1 were highly enriched in Ago2 groups (Fig. 5C), indicating that NR2F1-AS1 and NR2F1 coexisted in the RNA-induced silencing complex (RISC). By means of searching on starBase (http://starbase.sysu.edu.cn/), we discovered 3 miRNAs (miR-423-5p, miR-642a-3p, and miR-642b-3p) that harbored binding sites with both NR2F1-AS1 and NR2F1 (Fig. 5D). We then tested the expression levels of miR-423-5p, miR-642a-3p, and miR-642b-3p in CC cell lines and normal cervical cells. As displayed in figure 5E, miR-642a-3p expression was markedly upregulated in CC cells. Therefore, miR-642a-3p was selected for further analysis.
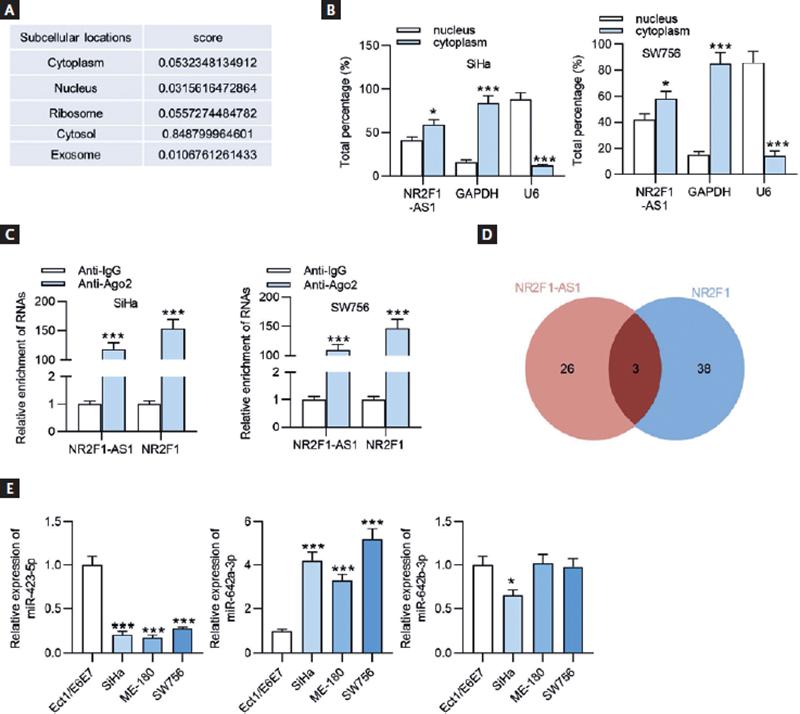
Figure 5. MiR-642a-3p is upregulated in CC cells. (A) The subcellular localization of NR2F1-AS1 was predicted by LncLocator database. (B) The subcellular localization of NR2F1-AS1 in SiHa and SW756 cell lines was validated by subcellular fractionation assay. GAPDH serves as the cytoplasmic localization control gene, and U6 serves as the nuclear localization control gene. (C) RIP assay was used to confirm the interaction between NR2F1-AS1 and NR2F1 in SiHa and SW756 cells. (D) The candidate miRNAs harboring binding sites with both NR2F1-AS1 and NR2F1 were predicted using starBase and shown as in Venn diagram. (E) The expression levels of 3 candidate miRNAs in CC cells and normal cervix epithelial cells (Ect1/E6E7) were assessed by RT-qPCR. *p < 0.05, ***p < 0.001.
NR2F1-AS1 functions as a molecular sponge for miR-642a-3p to upregulate NR2F1 expression
After transfection of miR-642a-3p mimics, miR-642a-3p expression level in SiHa and SW756 cells was significantly elevated (Fig. 6A). The results of RT-qPCR and Western blot showed that NR2F1 expression at both mRNA and protein levels was decreased on miR-642a-3p upregulation, while this effect was reversed after transfecting pcDNA3.1/NR2F1-AS1 (Fig. 6B). Subsequently, the binding site of miR-642a-3p on NR2F1-AS1 (chr5: 92745665-92745686[–]) or of miR-642a-3p on NR2F1 (chr5:92930105-92930133[+]) was predicted by using starBase online website (https://starbase.sysu.edu.cn/) (Fig. 6C). Luciferase reporter assay was conducted to validate the binding of miR-642a-3p on NR2F1-AS1 (or NR2F1). The results revealed that the luciferase activity of NR2F1-AS1-WT/NR2F1-WT plasmid was significantly attenuated by miR-642a-3p overexpression, while no difference was observed in NR2F1-AS1-Mut/NR2F1-Mut plasmid after transfecting miR-642a-3p mimics, indicating the combination of NR2F1-AS1 (or NR2F1) with miR-642a-3p in CC cells (Fig. 6D). In addition, RIP assay further demonstrated that NR2F1-AS1, miR-642a-3p, and NR2F1 were remarkably enriched in the beads conjugated with Ago2 antibody, indicating their coexistence in RISCs (Fig. 6E). Conclusively, NR2F1-AS1 acted as a ceRNA against miR-642a-3p to elevate NR2F1 expression.
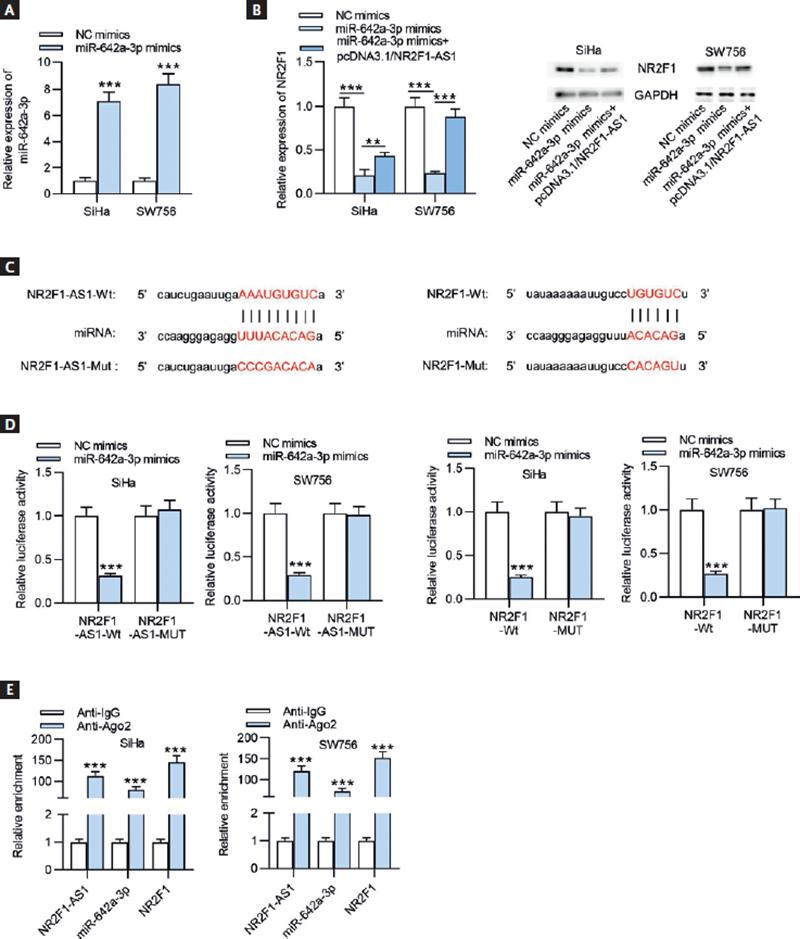
Figure 6. NR2F1-AS1 absorbs miR-642a-3p to upregulate NR2F1 expression. (A) The overexpression efficacy of miR-642a-3p in SiHa and SW756 cells was determined by RT-qPCR. (B) RT-qPCR and Western blot analysis were conducted to detect the mRNA and protein expression levels of NR2F1 in SiHa and SW756 cells transfected with NC mimics, miR-642a-3p mimics, and miR-642a-3p mimics+pcDNA3.1/NR2F1-AS1. (C) The binding sites of miR-642a-3p and NR2F1-AS1 (or NR2F1) were obtained from starBase. (D) The combination between miR-642a-3p and NR2F1-AS1 (or NR2F1) was verified through luciferase reporter assay. (E) RIP assay was performed to verify the coexistence of NR2F1-AS1, miR-642a-3p and NR2F1 in RISCs. **p < 0.01, ***p < 0.001.
NR2F1-AS1 suppresses CC cell malignant phenotypes by modulating the miR-642a-3p/NR2F1 axis
To explore the potential functional mechanism of NR2F1-AS1 in CC, rescue assays were designed. The mRNA and protein expression of NR2F1 was downregulated for the following experiments by transfecting SiHa cells with sh-NR2F1 (Fig. 7A). As demonstrated in figures 7B and C, NR2F1 knockdown or miR-642a-3p upregulation countervailed the inhibeffects mpacts of NR2F1-AS1 overexpression on CC cell proliferation. Upregulated NR2F1-AS1 suppressed cell migratory and invasive capacities, which were then antagonized by transfecting sh-NR2F1#2 or miR-642a-3p mimics (Fig. 7D and E). Through Western blot analysis and RT-qPCR, we found that NR2F1 knockdown or miR-642a-3p overexpression counteracted NR2F1-AS1 overexpression-mediated inhibition on EMT process in CC cells (Fig. 7F and G). Our data revealed that NR2F1-AS1 inhibited the malignant properties of CC cells by modulating the miR-642a-3p/NR2F1 regulatory axis.
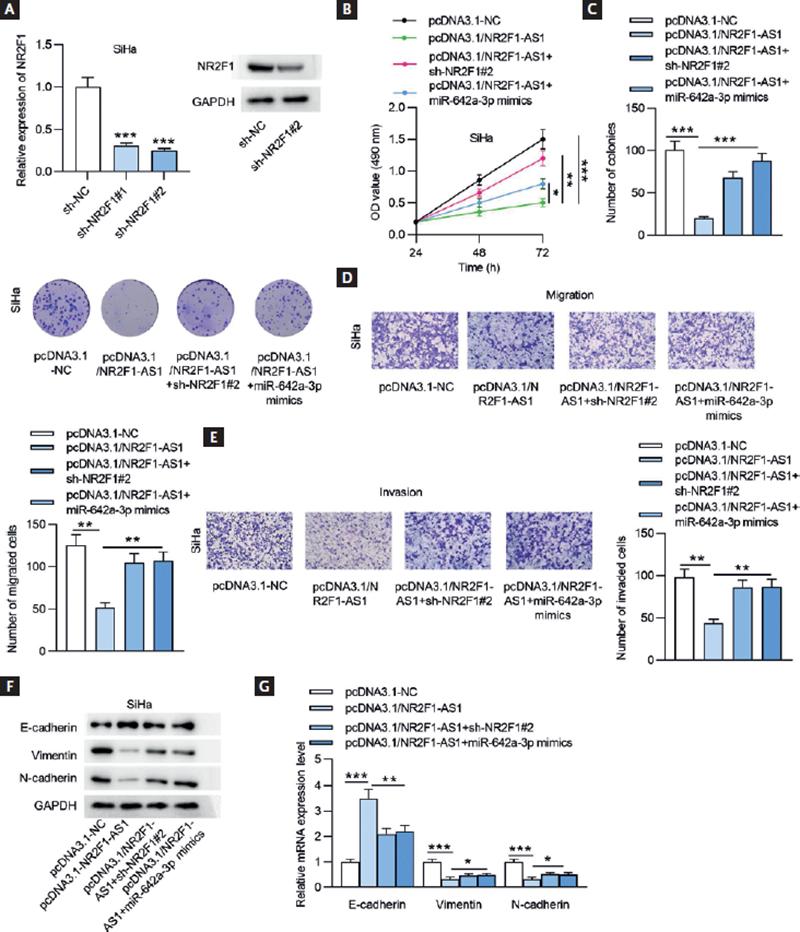
Figure 7. NR2F1-AS1 inhibits CC cell phenotypes by regulating the miR-642a-3p/NR2F1 axis. (A) The mRNA and protein expression of NR2F1 in SiHa cells transfected with sh-NR2F1 was detected by RT-qPCR and Western blot analysis. (B) MTT assay was conducted to examine cell viability after transfection with pcDNA3.1-NC, pcDNA3.1/NR2F1-AS1, pcDNA3.1/NR2F1-AS1+sh-NR2F1#2, pcDNA3.1/NR2F1-AS1+sh-NR2F#2, and pcDNA3.1/NR2F1-AS1+miR-642a-3p mimics. (C) Cell proliferation was estimated by performing colony formation experiment in different groups. (D-E) Transwell assays were performed to test the migratory and invasive capacities of CC cells after the indicated plasmids transfection. (F-G) The protein and mRNA expression levels of genes associated with EMT process were measured by Western blot analysis and RT-qPCR in SiHa cells after the transfection of the indicated plasmids. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Emerging evidence manifested that lncRNAs promote the biological activities in a variety of human cancers27-29. As is widely known, lncRNAs can affect cell phenotypes through different approaches30, one of which is the ceRNA network where lncRNAs may sequester miRNAs and thereby modulate the expression levels of mRNAs at the post-transcriptional level31. LncRNA NR2F1-AS1 has also been shown to actively participate in the cellular development of several human cancers21,32,33. Nonetheless, the biological function of NR2F1-AS1 in CC cells has not been fully elucidated. In our present study, we identified the downregulated expression of NR2F1-AS1 in CC tissues and cells. In addition, functional experiments demonstrated that NR2F1-AS1 upregulation repressed CC cell proliferation, migration, invasion, and EMT process. These findings demonstrated that NR2F1-AS1 acted as a tumor suppressor in CC, which is inconsistent with the general oncogenic role of NR2F1-AS1 in multiple other cancers, such as neuroblastoma34, gastric cancer35, and pancreatic cancer36.
Although multiple lncRNAs act in trans through RNA-RNA or RNA-protein interaction, emerging research has shown that some lncRNAs loci act locally (in cis) to modulate expression levels of nearby genes37. The previous studies have found that lncRNAs with high syntenic conserved characteristics across species are spatially correlated with neighboring transcription factors across the genome such as PTV1 and MYC, GATA6-AS1 and GATA6, LINC00261 and FOXA2, NR2F1-AS1 and NR2F138,39. Hence, we hypothesized that NR2F1 may exert an essential function in CC together with NR2F1-AS1. The previous works implied that NR2F1 was in close correlation with some cancers. For example, NR2F1 promotes cell proliferation and cell cycle process, but suppresses cell migration and invasion in salivary adenoid cystic carcinoma by increasing the expression of CXCL12 and CXCR4.40 Inhibition of NR2F1 facilitates early dissemination in breast cancer by inducing EMT process and a hybrid luminal/basal phenotype41,42. In our present study, we verified that NR2F1 displayed low levels in CC cells. Furthermore, restoration assays revealed that NR2F1 silencing rescued the biological behaviors of CC cells inhibited by upregulated NR2F1-AS1.
MicroRNAs (miRNAs) are short non-coding RNA molecules with nearly 22 nucleotides that exert essential functions in the post-transcriptional regulation of the expression of genes43,44. It is evident from the previous literature that miRNAs can target specific mRNAs to modulate the development of various cancers, including CC45. For example, miR-195-5p restrains CC cell migration and invasion by inhibiting ARL246. MiR-15a-5p targets TP53INP1 to obstruct CC progression47. Research has shown that lncRNAs could act as sponges of specific miRNAs to modulate the progression of human cancers48. LncRNA RBM5-AS1 binds with miR-1285-3p to facilitate oral squamous cell carcinoma invasive behaviors49. LncRNA NNT-AS1 influences breast cancer progression through miR-142-3p/ZEB1 axis.50 LncRNA LOXL1-AS1 serves as a ceRNA to decoy miR-423-5p and target MYBL2, thus facilitating the cellular progression of lung adenocarcinoma51. However, no evidence has revealed the molecular mechanism of miR-642a-3p in CC cells. Previously, Yu et al. demonstrated that miR-642a-3p downregulation enhanced chemosensitivity of resistant gastric carcinoma SGC7901 cells to 5-Fu by increasing FOXO4 expression52. MiR-642a represses hepatocellular carcinoma cell migration and invasion by targeting SEMA4C and then regulating p38 MAPK signaling pathway53. The present study showed that miR-642a-3p, as the downstream target of NR2F1-AS1, exhibited high expression level in CC cells. Mechanically, NR2F1-AS1 competitively combined with miR-642a-3p to modulate NR2F1 expression and thus regulate the malignant behaviors of CC cells.
In summary, our findings demonstrated that NR2F1-AS1 overexpression suppressed CC cell malignant phenotypes through adsorbing miR-642a-3p and targeting NR2F1, which might provide novel insights for exploring more potential molecular mechanisms related to CC.

