











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN

Las células deben coordinar su actividad metabólica con los cambios que sensan de su microambiente, para asegurar su crecimiento en condiciones favorables. Por lo tanto, para mantener la homeostasis celular debe existir una fina regulación entre la síntesis y la degradación de los componentes celulares. Los nutrientes, los factores de crecimiento y la energía activan vías biosintéticas y estimulan el crecimiento celular. Sin embargo, las células en respuesta a la limitación de nutrientes y otros estresores inducen una variedad de respuestas para favorecer su supervivencia, uno de estos mecanismos reguladores es la autofagia1.
El belga Christian de Duve acuñó por primera vez el término autofagia, para denominar a los procesos que realizan los lisosomas, y en 1974 ganó el premio Nobel de Medicina por sus hallazgos. La palabra autofagia proviene del griego “comerse a sí mismo” y tal ha sido su importancia fisiológica que en el año 2016 el japonés Yoshinori Ohsumi fue galardonado con el premio Nobel en Medicina por su investigación pionera en los genes relacionados con el proceso de autofagia (ATG, por sus siglas en inglés).
La autofagia es un mecanismo evolutivamente conservado entre diferentes especies, está presente a nivel basal en condiciones fisiológicas. Es la principal vía de remoción y degradación intracelular, en la cual se eliminan los componentes celulares senescentes o dañados, desde macromoléculas hasta organelos. Pero también es un control de calidad en donde las proteínas mal plegadas son degradadas. Así, la autofagia, dependiendo del contexto del microambiente, puede promover la supervivencia o la muerte celular2. La autofagia se activa en condiciones de limitación de nutrientes, ya que permite a la célula reutilizar sus propios componentes como nutrientes, contribuyendo a la movilización de distintos almacenes de energía como lípidos y carbohidratos, lo cual prolonga la vida de las células bajo estrés. La autofagia promueve la supervivencia celular no solo en la condición de privación de nutrientes, sino también por estrés del retículo endoplásmico (RE) o lisosomal o bien la hipoxia. Por el contrario, se ha documentado que en las células en condiciones donde existe un exceso de nutrientes, la autofagia se inhibe3. Más aún, la disminución en la actividad autofágica se ha asociado con la fisiopatología de enfermedades neurodegenerativas, cardiovasculares, cáncer y diabetes mellitus tipo 2 (DMT2)4-6.
AUTOFAGIA Y SUS MECANISMOS CELULARES

La autofagia es un proceso catabólico que puede clasificarse en: macroautofagia, microautofagia, autofagia mediada por chaperonas y autofagia selectiva. Estos se describen brevemente a continuación:
La macroautofagia, que a lo largo de esta revisión referiremos como autofagia, es un mecanismo vacuolar de autodigestión, responsable de la degradación vía lisosomal de más del 90% de las proteínas celulares, agregados macromoleculares y organelos dañados. En la macroautofagia, una pequeña parte del citoplasma es secuestrado por una membrana, la cual eventualmente forma una estructura de doble membrana denominada autofagosoma, ésta se fusiona con los lisosomas para formar el autofagolisosoma y el material secuestrado del citoplasma finalmente es degradado por las enzimas lisosomales7.
En la microautofagia, la membrana lisosomal directamente engulle pequeñas partes del citoplasma, inclusiones u organelos a degradar.
En la autofagia mediada por chaperonas los sustratos citoplasmáticos que contienen la secuencia de aminoácidos KFERQ son reconocidos por una proteína chaperona HSc70, la cual directamente los transloca al lisosoma para su degradación. A diferencia de la macroautofagia, en la microautofagia y en la autofagia mediada por chaperonas no se requiere la formación de vesículas intermediarias7.
Se ha denominado autofagia selectiva a aquella en la que las células autoingieren organelos específicos como ribosomas, lisosomas, mitocondrias, RE, proteasomas, peroxisomas, núcleos o lípidos y a dichos procesos autofágicos se les denomina como ribofagia, lisofagia, mitofagia, reticulofagia, proteofagia, pexofagia, nucleofagia o lipofagia, respectivamente8.
El proceso de autofagia es sumamente dinámico, los mecanismos celulares que participan en éste se han divididos en 3 fases consecutivas8, como se esquematiza en la figura 1.
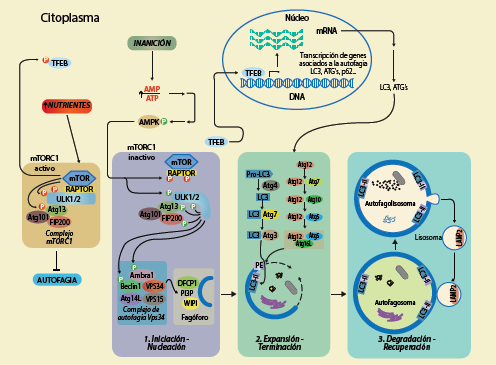
El complejo mTORC1 inhibe la autofagia en presencia de un exceso de nutrientes, fosforilando a ULK1 en sus residuos de serina 637 y 757 y a otras proteínas. En contraste, el aumento en la relación AMP/ATP durante la inanición conduce a la activación de la AMPK, misma que inactiva a mTORC1 fosforilando a RAPTOR y por ende se libera ULK1. AMPK a su vez activa a ULK1 fosforilándola en los residuos de serina 317 y 777 y ULK1 se fosforila a sí misma en el residuo 180 de treonina y a su vez fosforila a las proteínas Atg13, ATG101 y FIP200, dando inicio a las diferentes etapas de la autofagia.
Figura 1 Regulación de la autofagia en presencia o ausencia de nutrientes
Iniciación/nucleación. En condiciones de inanición, el aumento en la relación AMP/ATP conduce a la activación de la AMPK, misma que fosforila a RAPTOR dentro del complejo mTORC1, inhibiéndolo e iniciando así la autofagia. AMPK puede activar a la cinasa ULK1 mediante su fosforilación en los residuos de serina 317 y 777. También ULK1 puede activarse a sí misma fosforilándose en su residuo de serina 180. Una vez activa ULK1 es capaz de fosforilar a las proteínas Atg13, Atg101 y FIP2009. ULK1 enseguida fosforila a Ambra1, una proteína que interactúa con beclina1, la cual forma un complejo con Vps34, Vps15 y Atg14L. Vps34 es una proteína fosfatidil inositol 3-cinasa (PI3K, por sus siglas en inglés) que produce fosfatidil inositol 3-fosfato (PI3P) y recluta a las proteínas DFCP1, WIPI2 y proteínas ATG, iniciando la formación del fagóforo también denominado membrana de aislación8.
Expansión/terminación. Consiste de 2 vías semejantes a ubiquitina, en las que el sistema ATG es crítico, Atg7 actúa como una enzima parecida a E1 mientras que Atg10 y Atg3 actúan como una enzima E2. Atg12-Atg5-Atg6L1 actúan como un complejo enzimático parecido a E3. Por otro lado, Atg8 conocido como la proteína 1 de cadena ligera 3 asociada a microtúbulos (LC3, por sus siglas en inglés) se conjuga con Atg3 vía Atg7, luego LC3-Atg3 es reclutado a la membrana de aislación o fagóforo. Posteriormente, la proteína LC3 se conjuga a su blanco lipídico fosfatidiletanolamina (PE) en la membrana transformándose en LC3-II (la proteína LC3-II puede utilizarse como un marcador de autofagia temprana). Finalmente se completa la formación del autofagosoma, el cual puede identificarse mediante microscopía electrónica de transmisión ya que presenta doble membrana y contiene a los sustratos autofágicos intactos por la falta de enzimas proteolíticas, dado que aún no se ha fusionado con los lisosomas10. La proteína P62 también denominada SQSTM1, participa como una proteína adaptadora que enlaza proteínas ubiquitinizadas a LC3, de tal manera que cuando la autofagia es inhibida, P62 se acumula en las células.
Degradación/recuperación. Finalmente se fusionan el autofagosoma con el lisosoma, formando el autofagolisosoma, en donde las enzimas lisosomales inducen la degradación de los organelos y macromoléculas engullidas, completando así el proceso autofágico. Posterior a la degradación en el autofagolisosoma, los lisosomas se reciclan para volver a fusionarse con nuevos autofagosomas8, ver figura 1. Cabe mencionar que la proteína LAMP2 se localiza en las membranas lisosomales, y las catepsinas B y D representan a las principales proteasas lisosomales, de ahí que se utilizan como marcadores de la autofagia tardía10.
Como producto de la degradación, los nutrientes están disponibles y nuevamente mTORC1 se reactiva y forma el complejo con ULK1 inhibiendo de esta manera la autofagia. De lo anterior, el gen maestro regulador de la autofagia es mTOR (por sus siglas en inglés, mammalian target of rapamycin), este es capaz de sensar las fluctuaciones de los nutrientes en el microambiente para modular el crecimiento celular, el metabolismo y la supervivencia. mTOR es una proteína serina/treonina cinasa evolutivamente conservada, que puede formar 2 complejos: mTORC1 y mTORC2 que son estructural y funcionalmente diferentes. mTORC1 es el blanco farmacológico de la rapamicina, y mTORC2 puede llegar a ser sensible a ésta si se expone a ella por tiempos prolongados1. De ahí que en condiciones experimentales mTORC1 se puede inhibir con rapamicina, aumentando así la autofagia. En condiciones fisiológicas de privación de nutrientes mTORC1 es inhibido y por ende la autofagia incrementa. En contraste, la autofagia es regulada a la baja en los mamíferos cuando la cinasa mTORC1 es activada por un exceso de nutrientes11 (figura 1).
Río arriba de mTORC1 se encuentra la proteína cinasa-AMP (AMPK, por sus siglas en inglés), la cual responde a niveles reducidos de ATP y al consecuente incremento de AMP, fosforilando a mTORC1, lo cual lo inhibe y en respuesta se activa la autofagia12, como se muestra en la figura 1.
Además, en las células eucariotas, se ha descrito que la autofagia se regula transcripcionalmente por el factor de transcripción EB (TFEB, por sus siglas en inglés), considerado un gen maestro para la biogénesis lisosomal y su actividad se modula río arriba por mTORC1. Cuando en las células existe un exceso de nutrientes, mTORC1 fosforila a TFEB y éste permanece en el citosol. No obstante, cuando las células están en condición de inanición TFEB es desfosforilado y puede translocarse al núcleo, activando así la transcripción de genes lisosomales y ATGs los cuales participan activamente en la autofagia13, como se muestra en la figura 1,.
AUTOFAGIA EN LAS CÉLULAS BETA PANCREÁTICAS
Se ha reportado que la autofagia es el mayor regulador de la homeostasis de las células beta pancreáticas14. La autofagia juega un papel central en el mantenimiento de la masa y la función de las células beta. Su desregulación se ha asociado a obesidad, resistencia a la insulina e intolerancia a la glucosa, como lo demostró el modelo de ratón generado con una deleción específica en la célula beta para el gen ATG7, el cual es un gen esencial para la formación del autofagosoma, con lo cual se inhibió la autofagia. El ratón con la deleción ATG7 presentó hiperglucemia, disminución de la tolerancia a la glucosa, así como disminución en la masa de las células beta y en su contenido de insulina como consecuencia de un incremento en la apoptosis. Los resultados indican que la autofagia es indispensable para mantener la estructura, la masa y la función de las células beta pancreáticas15. La mitocondria y el RE son organelos que tienen un papel vital en la función y supervivencia de las células beta pancreáticas, la alteración de la autofagia para degradar estos organelos cuando están dañados puede inducir una disminución en la sensibilidad a la insulina en los tejidos blanco e incluso la muerte celular8.
Los tipos de autofagia que han sido actualmente investigados por su relevancia en el fallo de las células beta pancreáticas son: la mitofagia, la reticulofagia, la lipofagia y la crinofagia, los cuales se explican a continuación.

Mitofagia
La mitofagia es la autofagia selectiva de las mitocondrias, la cual sucede de manera basal como un mecanismo de mantenimiento y recambio de organelos, pero también se activa cuando se detecta estrés de la mitocondria16. En las células beta pancreáticas la mitofagia tiene una gran relevancia debido al papel que la mitocondria realiza, este organelo es crítico en el mantenimiento del equilibrio energético y la protección contra el estrés oxidativo. Las mitocondrias producen abundantes radicales de oxígeno y son particularmente susceptibles al daño de las especies reactivas de oxígeno (ROS, por sus siglas en inglés), cuando se pierde el equilibrio en los sistemas antioxidantes. Frente a un daño mitocondrial producido por estrés oxidante, el complejo mTOR se inactiva para dar paso a la mitofagia, en la cual la mitocondria cambia su potencial de membrana y se despolariza promoviendo la unión de varias proteínas cinasas PINK1 en la membrana externa de la mitocondria. PINK1 luego recluta a una ubiquitina-proteína ligasa E3 citosólica denominada PARK2/Parkin, que regula la ubiquitinación de proteínas mitocondriales, las cuales son reconocidas por las proteínas adaptadoras P62 y HDAC6, mismas que las unen a LC3, iniciando la degradación de la mitocondria8 Existe otro proceso que induce la mitofagia y que involucra a la proteína NIX la cual presenta un dominio de interacción con LC3, lo que permite el englobamiento de mitocondrias marcadas con NIX en autofagosomas17.
Reticulofagia
Esta juega un papel muy importante para el buen funcionamiento celular, el RE debe mantenerse en óptimas condiciones para asegurar el correcto pleglamiento de las proteínas. La síntesis de insulina representa el 50% de la producción proteica total de las células beta del páncreas. De tal forma, que un aumento en la ingesta calórica exige incrementar la síntesis y la secreción de insulina a fin de mantener la homeostasis metabólica de las células beta. El estrés del RE puede provocar un defecto en el plegamiento de proteínas que incluyen a la insulina. La respuesta celular a proteínas no plegadas (UPR, por sus siglas en inglés), es un proceso biológico esencial que tiene el objetivo de disminuir la sobrecarga de proteínas mal plegadas al RE, por el que las células intentan recuperar la homeostasis del mismo. Una serie de respuestas para disminuir la entrada de proteínas mal plegadas al RE incluyen un aumento en la degradación del ARN mensajero, la reducción en la traducción y la degradación de proteínas mediada por el proteasoma. Sin embargo, la reticulofagia es el principal mecanismo para la degradación de proteínas intracelulares mal plegadas18. El proceso de reticulofagia para la renovación y el buen funcionamiento del RE es crucial en las células beta. El estrés oxidante del RE puede darse en condiciones fisiológicas, como sucede en la célula beta con una alta carga biosintética sufrida tras la estimulación postprandial de síntesis de insulina. El complejo mTOR se inactiva para dar paso a la reticulofagia, para ello, la proteína FAM134B se une a la membrana del mismo e interactúa con LC3 para promover la formación del autofagosoma englobando al RE para su posterior degradación a través de la fusión con el lisosoma y la actividad de las enzimas lisosomales8.
Lipofagia
Recientemente se han investigado los mecanismos celulares involucrados en la lipofagia, la cual se define como la degradación autofágica de las gotitas de lípidos intracelulares. Se han descrito 3 subtipos de lipofagia19:
Lipofagia asociada a chaperona, en esta participan las proteínas de la familia de las peripilinas, las cuales identifican un dominio (KFERQ) sobre la chaperona HSC70. Una vez unida va hacia el lisosoma e interactúa con una proteína de membrana denominada LAMP2 dimerizada que transloca a la peripilina y ésta es la señal que promoverá la lipofagia de la gota lipídica.
La macrolipofagia, es mediada por la activación de la vía de señalización que transloca TFEB al núcleo e induce la trascripción de genes lisosomales y ATGs, lo cual culmina con la formación del autofagosoma, fusión y degradación de la gota lipídica por las enzimas lisosomales.
La microlipofagia, consiste en engullir directamente la gota lipídica por el lisosoma para posteriormente ser degradadas por las lipasas lisosomales19.
Crinofagia
El término crinofagia se refiere a la degradación de vesículas de secreción, proceso que ocurre directamente por la fusión de los gránulos secretorios con los lisosomas12. En el caso particular de las células beta, la crinofagia es un tipo de autofagia utilizada para la degradación de la insulina cuando las células se encuentran tanto en estado de ayuno temprano20 como prolongado21. En condiciones fisiológicas normales, las células beta pancreáticas mediante la proteína cinasa D (PKD, por sus siglas en inglés) inactiva, mantienen inhibido el sistema de degradación de gránulos nacientes inducida por estrés (SINGD, por sus siglas en inglés). En estado de ayuno, cuando no se requiere la presencia de insulina en las células beta se induce la crinofagia, se activa la PKD y, por ende, se desinhibe al SINGD. Los aminoácidos liberados de la degradación de la insulina son reutilizados por la maquinaria celular y mantienen la homeostasis de las células beta8.
De manera general, la regulación de la autofagia en las células beta pancreáticas depende de las señales del microambiente, como se muestra en la figura 2, donde los inductores de la autofagia son la inanición e hipoglucemia y por ende la disminución en los nutrientes, así como el estrés oxidativo de las mitocondrias provocado por un aumento en la producción de ROS o el estrés del RE derivado de las proteínas no plegadas, en cuyo caso el complejo mTORC1 se inhibe y la autofagia tiene lugar22. Lo anterior permite descartar los organelos dañados o citotóxicos como en el caso de los lípidos y mantener así la homeostasis de la célula beta y por lo tanto favorece su supervivencia. Por otro lado, los inhibidores de la autofagia en las células beta son todos aquellos que mantienen en modo activo al complejo mTORC1 (exceso de nutrientes, hiperglucemia, hiperlipidemia, una concentración elevada de ATP), lo cual detiene la autofagia. Existe consenso por distintos autores que la inhibición de la autofagia causa la apoptosis de las células beta pancreáticas23,24, lo cual sugiere que la autofagia funciona como un mecanismo protector para la supervivencia de las células beta. La muerte de las células beta lleva a la pérdida de la masa de las células beta en el islote pancreático, un rasgo clínico característico de la DMT2.
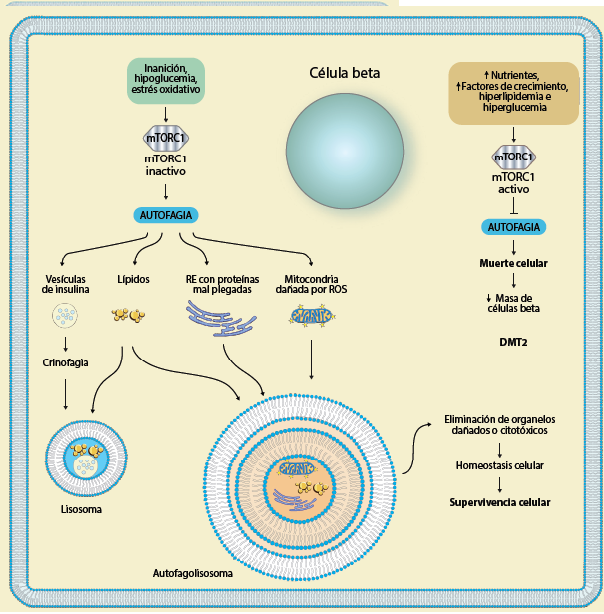
mTORC1 es un regulador clave de la autofagia, el cual es capaz de sensar el nivel de nutrientes en el microambiente. Por lo tanto, la inanición, la hipoglucemia y el estrés oxidativo desencadenan la autofagia, lo que permite conservar la homeostasis y supervivencia de las células beta. En contraparte, se inhibe la autofagia cuando mTORC1 detecta que hay un exceso de nutrientes. Si esto persiste de manera crónica las células beta mueren y ello conduce a la pérdida en la masa de las células secretoras de insulina, contribuyendo al desarrollo de la DMT2.
Figura 2 Autofagia en las células beta pancreáticas en respuesta al microambiente
AUTOFAGIA Y GLUCOLIPOTOXICIDAD
Las células beta del páncreas funcionan como sensores de nutrientes que regulan la secreción de insulina en respuesta a niveles elevados de glucosa y de lípidos. La sobrecarga de nutrientes es conocida como la principal causa de resistencia a la insulina, ya que incrementa la demanda de secreción de insulina por estas células25. Así, el deterioro de la función y la supervivencia de las células beta pancreáticas ocasionado por la exposición a concentraciones suprafisiológicas de glucosa se denomina glucotoxicidad, la cual se ha asociado con la inducción de estrés del RE, disfunción mitocondrial y daño oxidativo a proteínas26. Una dieta alta en glucosa se ha asociado con el desarrollo de enfermedades metabólicas como obesidad y DMT2. La glucosa en exceso crónico causa efectos tóxicos sobre la estructura y función de diversos órganos, incluyendo a los islotes pancreáticos de rata y de humano, induciendo apoptosis en las células beta pancreáticas27,28. Más aún, se ha descrito que en los islotes pancreáticos de humano y en líneas celulares productoras de insulina (INS-1 y MIN6) expuestas a niveles elevados de glucosa durante 16 horas se bloquea el flujo autofágico, resultando en una acumulación de autofagosomas, lo cual conduce a dichas células a la muerte celular apoptótica10. En otro estudio reportaron que las células INS-1 fueron incubadas durante 48 horas con una concentración alta de glucosa, encontrando una inhibición de la translocación de TFEB al núcleo y por ende una disminución en los transcritos de Lamp1, P62 y LC325. De lo anterior existe consenso en que una dieta elevada en glucosa provoca una disminución en la autofagia, con el consecuente deterioro de la célula beta pancreática.
Respecto a los ácidos grasos (AG), estos se clasifican con base en la longitud de la cadena de carbonos y por la presencia de uno o más dobles enlaces. De lo anterior, pueden clasificarse en insaturados (ácido oleico), saturados (ácido palmítico), monoinsaturados (ácido palmitoleico) o poliinsaturados (ácido linoleico)29,30. Los AG están involucrados en el metabolismo energético de la mayoría de los organismos y son conocidos por regular el equilibrio de las células beta pancreáticas. Estos cumplen funciones como la formación de moléculas señalizadoras de naturaleza lipídica, la producción de energía a través de la β-oxidación, la modificación postraduccional de proteínas, así como la regulación de la transcripción y la biosíntesis de membranas celulares18. Los AG, presentan diferentes vías metabólicas, estos pueden ser destinados a la formación de lipoproteínas y fosfolípidos o ser reesterificados para almacenarse en forma de triglicéridos. La reesterificación de los AG requiere la activación de la acil CoA y la reacción genera un aumento de AMP, éste a su vez activa a la AMPK, la cual:
Promueve la β-oxidación de los AG para generar energía o cuerpos cetónicos (con ello se evita la lipotoxicidad).
Reduce la lipolisis.
Es capaz de inducir procesos de autofagia.
Cabe señalar que el ácido oleico y el ácido palmítico son los AG predominantes en la circulación sanguínea y ambos aumentan cuando se consumen dietas altas en grasa31. Se ha observado que el incremento crónico en los niveles circulantes de AG daña la secreción de insulina estimulada por glucosa, induce resistencia a la insulina y disfunción de la célula beta pancreática, dichos efectos deletéreos de los AG sobre la homeostasis de la glucosa son referidos como lipotoxicidad30. Otros mecanismos considerados como mediadores de la lipotoxicidad en las células beta son el estrés del RE ocasionado por el aumento en la concentración de ácido palmítico y el estrés oxidante. Dicho estrés oxidante es ocasionado por la β-oxidación de los AG, lo que produce un incremento de las ROS. De no disminuir el estrés en las células beta se induce la muerte celular apoptótica18. La alteración en la regulación del metabolismo de lípidos también se ha asociado con obesidad, siendo uno de los principales factores que influyen en el incremento en la incidencia de DMT28.
Es importante destacar que los efectos de los AG dependen de la longitud y grado de saturación de la cadena carbonada18, como lo evidencian distintos estudios. Por ejemplo, es sabido que los AG saturados son particularmente citotóxicos para las células beta, induciendo la apoptosis, en tanto que los AG insaturados protegen a las células de la apoptosis32.
En un estudio realizado en islotes pancreáticos de rata incubados durante 4 días con ácido palmítico (0.5 mM) o ácido palmitoleico (0.5 mM), se observó que el ácido palmítico redujo la capacidad proliferativa de las células beta e indujo la muerte celular por apoptosis. Por el contrario, el ácido palmitoleico, promovió la proliferación de las células beta, contrarrestando los efectos citotóxicos del ácido palmítico33.
Se ha reportado que las concentraciones elevadas de AG saturados inhiben la autofagia en las células beta pancreáticas, como se evidenció en estudios in vitro con islotes pancreáticos humanos incubados con ácido palmítico (0.4 mM) durante 48 horas, observando una detención en el flujo autofágico10. Consistentemente en células INS-1E expuestas durante 18 horas con ácido palmítico (0.25 mM), se observó que este detiene el flujo autofágico y disminuye la viabilidad celular. En contraste, cuando estas células se coincuban con ácido palmítico y ácido palmitoleico se observó un aumento en la viabilidad celular y en la actividad autofágica, lo que sugiere que el ácido palmitoleico puede tener una propiedad protectora frente a los efectos tóxicos del ácido palmítico34.
Sin embargo, cuando las células INS-1 se incuban durante 24 horas con ácido oleico (0.5 mM), éste es capaz de inducir la autofagia. Lo que sugiere que la autofagia actúa como un mecanismo adaptativo de las células beta en respuesta a la resistencia a la insulina inducida por la dieta alta en grasa saturada35, ya que los AG insaturados si promueven el proceso de autofagia.
La exposición crónica a concentraciones de glucosa y AG elevados presentan un efecto sinérgico, denominado glucolipotoxicidad30. Choi y colaboradores incubaron las células INS-1 durante 12 horas con ácido palmítico (0.4 mM) en presencia de glucosa (25 mM), observando un aumento en la autofagia a través de la formación de vesículas autofágicas. Una posible explicación en este modelo es que la autofagia se active como una respuesta adaptativa ante el estrés metabólico, aunado al hecho que el tiempo de exposición a la glucolipotoxicidad es relativamente corto32. Contrariamente en un estudio en el que tanto las células INS-1 como islotes pancreáticos humanos fueron expuestos durante aproximadamente 18 horas a niveles elevados de glucosa (30 mM) se indujo su apoptosis. Sin embargo, cuando se incubaron durante 16 horas con alta concentración de glucosa en combinación con ácido palmítico (0.4 mM) se indujo un mayor incremento en la muerte celular apoptótica. Los autores mostraron que, la sobrecarga de nutrientes resultó en una disminución de la autofagia con incremento en la acumulación de autofagosomas, estrés del RE, acumulación de mitocondrias aberrantes, disfunción lisosomal y activación de mTORC1, lo cual condujo a la muerte a las células beta pancreáticas10. Lo anterior demuestra que la glucosa y los lípidos en concentraciones elevadas durante tiempos prolongados inhiben la autofagia y ello conduce a la muerte celular, lo que favorece el desarrollo de enfermedades metabólicas tales como obesidad y diabetes. Distintas investigaciones han hecho patentes los efectos de la glucolipotoxicidad en la autofagia de las células beta del páncreas, algunos de los cuales se resumen en la tabla 1.
Tabla 1 Efectos de la glucolipotoxicidad en la autofagia de las células beta pancreáticas
| Modelo experimental | Concentración de glucosa, lípidos o ambos | Tiempo de exposición | Efecto observado | Cita |
|---|---|---|---|---|
| Células INS-1, MIN6 e islotes pancreáticos humanos | Glucosa 33.3 mM | 16 horas | Bloqueó del flujo autofágico e incremento de la apoptosis de las células secretoras de insulina. | 10 |
| Islotes pancreáticos de ratones C57BL/6 | Glucosa al 10% en agua | 12 semanas | No se observó inducción de la autofagia vía la conversión de LC3 a LC3-II y mediante MET raramente se observaron vacuolas autofágicas | 53 |
| Células INS-1 | Glucosa 30 mM | 48 horas | Se inhibe la translocación de TFEB al núcleo, y disminuyen los transcritos de Lamp1, P62 y LC3. En resumen, la autofagia disminuye | 25 |
| Células INS-1 | Ácido oleico 0.5 mM | 24 horas | Se observó un mayor flujo autofágico a través de la conversión de LC3I a LC3II | 35 |
| Células MIN6 | Ácido palmítico 0.5 mM | 36 horas | Incremento de la autofagia como un mecanismo protector del estrés del RE ocasionado por el ácido palmítico | 54 |
| Islotes de ratones C57BL/6 | Dieta alta en grasa (40.6%) para inducir diabetes ligada a obesidad | 12 semanas | Flujo autofágico detenido y muerte celular | 55 |
| Células INS-1E | Ácido palmítico 0.25 mM | 18 horas | Detención del flujo autofágico y disminución de la viabilidad celular | 34 |
| Islotes pancreáticos de ratones cepa C57BL/6 y células MIN6 | Ácido oléico 0.4mM o ácido palmítico 0.2 mM | 48 horas | Comparado con el ácido palmítico el ácido oleico aumentó el flujo autofágico, incrementando la fusión del autofagosoma con el lisosoma | 31 |
| Células INS-1 | Ácido palmítico 0.4 mM en presencia de glucosa 25 mM | 12 horas | Aumento de la autofagia, se observó la formación de vesículas autofágicas y disminución de la muerte celular por apoptosis | 32 |
| Células INS-1 832/13 | Ácido palmítico 0.4 mM en presencia de glucosa 20 mM | 22 horas | Disminución del flujo autofágico | 56 |
| Células INS-1E e islotes pancreáticos humanos | Ácido palmítico 0.5 mM en presencia de glucosa 25 mM | 16 horas | Bloqueo del flujo autofágico asociado con disfunción lisosomal y muerte celular | 50 |
AUTOFAGIA Y DMT2
De acuerdo con la Organización Mundial de la Salud actualmente hay 422 millones de personas con diabetes y más del 80% de las muertes ocasionadas por esta enfermedad se registran en países de ingresos bajos a medios36. En el año 2020, la Federación Mexicana de Diabetes reportó que la DMT2 es la segunda causa de mortalidad en México. Más aún, México es el segundo país latinoamericano y el sexto del mundo en prevalencia de este trastorno con casi 11.5 millones de pacientes37.
La DMT2 constituye alrededor del 95% de todos los casos de diabetes mellitus y su patogénesis es un proceso complejo que depende completamente del estado funcional de las células beta pancreáticas secretoras de insulina. La DMT2 es considerada como un conjunto de alteraciones metabólicas. A menudo tiene un inicio en la edad adulta y se caracteriza por una hiperglucemia en ayunas moderada, asociada con una marcada resistencia a la insulina, la cual es parcialmente compensada por hiperplasia de las células beta. Posteriormente, se caracteriza por una hiperglucemia crónica, debida a la pérdida progresiva de la secreción de insulina, asociada a resistencia a la insulina, además de que los pacientes presentan también niveles elevados de ácidos grasos. A su vez la hiperinsulinemia se origina como una consecuencia compensatoria a la resistencia a la insulina. Finalmente, la muerte de las células beta pancreáticas produce la pérdida de la masa de las células beta en los islotes pancreáticos18.
En pacientes con DMT2, la hiperglucemia potencia la generación excesiva de ROS derivando a un estrés oxidativo38. El islote pancreático es especialmente vulnerable a las ROS debido a su bajo nivel intrínseco de enzimas antioxidantes26, las mitocondrias y el RE son susceptibles al daño por ROS y son organelos clave para la homeostasis de las células beta8. El estrés oxidativo está estrechamente asociado con la inflamación crónica que se presenta en la DMT2. El inflamosoma es un complejo de proteínas que actúan como sensores para patrones moleculares asociados a patógenos (PAMPs, por sus siglas en inglés). Las concentraciones elevadas de ROS son detectadas por el inflamosoma como un PAMP, desencadenando la piroptosis, un tipo de muerte celular que se caracteriza por ser proinflamatoria. El mecanismo de ejecución es a través de la activación de la caspasa 1, la cual a su vez activa a la IL-1β, generando edema, la formación de un poro en la membrana y finalmente la lisis celular. La autofagia parece proteger a las células de la piroptosis, degradando a las mitocondrias dañadas y componentes del inflamosoma, limitando así la producción de ROS18.
También se ha descrito que la muerte de las células beta pancreáticas se debe en parte a los niveles elevados de amilina, también denominada polipéptido amiloide (IAPP, por sus siglas en inglés). El IAPP es una hormona formada por 37 aminoácidos, en los humanos es sintetizada, almacenada y cosecretada con la insulina, por lo que en la DMT2 la secreción excesiva de insulina causada por la resistencia a la insulina en tejidos periféricos hace que el IAPP también se encuentre en altas concentraciones locales, provocando la formación de depósitos amiloides39. Dichos depósitos forman oligómeros de amilina tóxicos, que junto con el heparán sulfato y los glucosaminoglicanos forman una malla. Al proceso donde se depositan precipitados insolubles de amilina (fibrillas), se conoce como amiloidogénesis40. Lo anterior es consistente con lo reportado en autopsias de pacientes con DMT2, donde han observado una acumulación de fibras amiloides en los islotes pancreáticos. Los mecanismos por los cuales el IAPP puede llevar a la muerte a las células beta son varios. Por un lado, los agregados de IAPP generan estrés, activación del inflamosoma y piroptosis. Por otra parte, el IAPP deteriora la autofagia como lo demuestra la acumulación de P62 y la disminución de la autofagia conduce a una mayor acumulación del IAPP y muerte celular de los islotes pancreáticos8. La apoptosis ha sido considerada como el principal mecanismo de muerte de las células beta pancreáticas cuando la autofagia se encuentra inhibida, como ha evidenciado el modelo de ratón deficiente para el gen ATG7, el cual mostró disminución en la masa de las células beta y un aumento significativo de apoptosis10.
Finalmente, existe consenso por diferentes autores, que la disminución en la autofagia en las células beta pancreáticas contribuye al desarrollo de la DMT28,41-44.
FÁRMACOS REGULADORES DE LA AUTOFAGIA
Durante la patogénesis de la DMT2 el exceso de AG y glucosa generan condiciones de glucolipotoxicidad, lo cual inhibe la autofagia en las células beta pancreáticas y las conduce a la muerte30. De lo anterior, si la disminución de la autofagia en las células beta se asocia con el desarrollo de la DMT2, el tratamiento con fármacos u otros agentes químicos que estimulen la autofagia podría ser una nueva estrategia clínica para tratar o prevenir la diabetes.
Se sabe que múltiples factores estimulan la autofagia en las células beta del páncreas de pacientes con DMT2, estos incluyen algunos componentes dietéticos, citocinas, fármacos y agentes químicos. Se ha observado que el ayuno prolongado y la dieta restringida en calorías estimulan la autofagia tanto in vitro como in vivo. Por otra parte, se ha identificado en frutas y verduras el kaempferol, un flavonol inductor de la autofagia. También se ha mostrado que los AG omega-3 encontrados en el salmón, aumentan la autofagia en ratones a los cuales se les indujo diabetes con estreptozotocina. Estos hallazgos sugieren que los nutrientes o antioxidantes específicos presentes en los alimentos podrían usarse selectivamente para estimular la autofagia de las células beta. También, se ha observado que ciertas citocinas juegan un papel protector de las células beta pancreáticas, la IL-22 e IL-6 estimulan la autofagia y promueve la supervivencia de las células protegiéndolas de la apoptosis inducida por el TNF-α, la IL-1β e IFN-ϒ45.
La metformina, la glibenclamida, la rosiglitazona o el péptido 1 similar al glucagón (GLP-1, por sus siglas en inglés), son tratamientos usados en la DMT2 y estos han demostrado estimular la autofagia en las células beta y prevenir la apoptosis en condiciones de glucolipotoxicidad30,46.
En pacientes diabéticos se ha observado un incremento en el contenido de autofagosomas, lo cual puede deberse a un bloqueo en la autofagia. El tratamiento con metformina reduce el contenido de autofagosomas probablemente a través de la activación de la AMPK, aumentando así la autofagia8. Otros autores han reportado que la metformina estimula la autofagia de las células beta y evita la apoptosis bajo condiciones de lipotoxicidad in vitro47.
Zhou y colaboradores, demostraron en estudios in vitro que la glibenclamida induce la autofagia en las células MIN6. Ellos determinaron que la activación de la AMPK produce un aumento significativo en los marcadores CL3-I y CL3-II, los cuales son indicadores de la actividad autofágica48.
La rosiglitazona es un fármaco que pertenece a la familia de las tiazolidinedionas y también se emplea en el tratamiento de la DMT2. En un estudio realizado in vitro en las células INS-1 se observó que el ácido palmítico en altas concentraciones provoca la apoptosis de estas células. Sin embargo, cuando las células se incubaron previamente con rosiglitazona y posteriormente con el ácido palmítico, se observó una disminución en la apoptosis debida al incremento en la autofagia. El efecto protector de la rosiglitazona se debió al aumento de la autofagia vía la activación de la AMPK49.
Recientemente se demostró que GLP-1 es capaz de estimular la autofagia. Las células INS-1E e islotes pancreáticos humanos, fueron incubados durante 16 horas en condiciones de glucolipotoxicidad con ácido palmítico (0.5 mM) y glucosa (25 mM), observando en estos una inhibición de la autofagia y un aumento en la muerte celular. Sin embargo, cuando las células INS-1E y los islotes fueron incubados bajo las mismas condiciones de glucolipotoxicidad más exendina-4, un agonista de GLP-1, se observó que se restaura la función lisosomal, se incrementa el flujo autofágico y la supervivencia celular50.
Adicional a los fármacos usados para el tratamiento de la DMT2, algunos agentes químicos pueden mejorar el perfil metabólico de animales diabéticos o con síndrome metabólico incrementando la actividad autofágica, como se describe a continuación.
Como se explicó antes, el modo inactivo de mTORC1 estimula la autofagia, de lo anterior existe un gran interés en encontrar fármacos que bloqueen a mTORC1, con la meta de que en un futuro sean implementados como parte del tratamiento para la DMT2. Se ha demostrado que la rapamicina estimula la autofagia de las células beta in vitro e in vivo, pero hay informes contradictorios sobre sus efectos colaterales, ya que el tratamiento sostenido con rapamicina resultó en una disminución de la función y viabilidad de las células beta. Otros investigadores demostraron que el tratamiento intermitente con rapamicina, elimina los efectos metabólicos negativos del tratamiento diario con esta. De lo anterior, es necesario realizar un número mayor de estudios con rapamicina para poder asegurar su eficacia en el tratamiento de la DMT2 a través del aumento en la autofagia45.
Hyejin y colaboradores sintetizaron una molécula química a la cual denominaron “MSL” y demostraron que es capaz de aumentar la autofagia en el modelo de ratones obesos, de la cepa ob/ob de Jackson. A estos ratones de 8 semanas de edad les administraron MSL 3 veces a la semana durante 8 semanas, observando una disminución en el nivel de glucosa sanguínea, una mejoría en la tolerancia a la glucosa y en la sensibilidad a la insulina. Todo lo anterior acompañado por un aumento en el flujo autofágico. Interesantemente, encontraron que el mecanismo bajo el cual la molécula MSL aumenta la autofagia es a través de la calcineurina, la cual desfosforila a TFEB y éste consigue translocarse al núcleo y estimular así la autofagia51.
Por otro lado, en un modelo de ratones de la cepa C57BL/6 de 10 semanas de edad Tuohetaerbaike y colaboradores indujeron diabetes mediante una dieta alta en grasa más dosis bajas de estreptozotocina. Posteriormente investigaron el efecto protector antidiabético de la urolitina A administrándola durante 8 semanas a estos ratones. Los autores observaron una disminución de la glucosa sanguínea en ayunas, un aumento de la tolerancia de la glucosa, y disminución en las citocinas inflamatorias. Se observó que la urolitina A indujo un incremento en la expresión de proteínas involucradas en la autofagia como beclina 1, ATG5 y LC3II, así como una disminución de P62. En conclusión, la urolitina A incrementó la autofagia en las células beta de los ratones diabéticos y disminuyó su apoptosis52.
CONCLUSIONES
Existe consenso en las distintas investigaciones tanto in vitro como in vivo que la sobrecarga de nutrientes disminuye la autofagia en las células beta pancreáticas y en consecuencia incrementa la apoptosis de estas células, lo cual contribuye al desarrollo de la DMT2. Los estudios realizados en modelos de roedores obesos o diabéticos tratados con fármacos o agentes químicos que promueven la autofagia, han permitido observar una mejoría en la viabilidad de las células beta y una disminución en la resistencia a la insulina. Lo anterior resulta prometedor para ser trasladado al tratamiento de pacientes con DMT2 o con síndrome metabólico en un futuro cercano. Sin embargo, los resultados observados deben ser considerados con precaución, dado que algunos son estudios realizados in vitro y en modelos animales, los cuales podrían no reflejar exactamente los mismos resultados en pacientes humanos con diabetes. Así como también se deben realizar investigaciones con agentes potenciadores de la autofagia dirigidos específicamente a las células beta pancreáticas. De lo anterior, hacen falta más estudios para el desarrollo de nuevas terapias para los pacientes con DMT2 que justifiquen el uso de estos potenciadores de la autofagia.
REFLEXIONES
Aunada a esta propuesta de investigación farmacológica, es necesario hacer una profunda reflexión acerca del estilo de vida de la población, con la meta de prevenir enfermedades crónico metabólicas. Es fundamental que las personas procuren hábitos más saludables, que incluyan una dieta integral, una rutina diaria de ejercicio y una disminución del estrés. Más aún, se requiere de iniciativas de políticas gubernamentales que regulen a la industria de alimentos para generar productos con menores contenidos de azúcares y grasas, así como a concientizar a la población para disminuir sus hábitos de consumo.

