












 (pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
PermalinkIntroducción
El carcinoma de células de Merkel (CCM) es un carcinoma neuroendocrino primario de la piel poco frecuente1,2. Afecta predominantemente a personas mayores, caucásicas, y presenta un comportamiento agresivo, con una alta tasa de recurrencia y propensión a la metástasis temprana3,4.
A pesar de los nuevos avances en las terapias para el CCM, el pronóstico sigue siendo desalentador5. Por ello es de suma importancia el diagnóstico precoz y un tratamiento eficaz para mejorar la atención al paciente y mejorar la sobrevida6,7.
Ya que al no contar con el arsenal completo de marcadores puede pasar desapercibida la enfermedad y erróneamente catalogarla como principal diagnóstico diferencial con un sarcoma de tejidos blandos, y al tratarse de un cáncer raro y agresivo de piel, el tratamiento eficaz aumenta la sobrevida y el periodo libre de recurrencia, es por ello la importancia de presentar el caso8.
Reporte de caso
Paciente de sexo femenino de 79 años, con antecedentes de depresión mayor en tratamiento. Antecedentes heredofamiliares: refiere hermano finado por cáncer no especificado y sin referir antecedentes oncológicos por el paciente. Antecedentes no patológicos: dedicada al hogar, toxicomanías negadas, esquema de vacunación acorde a edad y completo.
Comienza su padecimiento en marzo del 2023 con dermatosis de glúteo izquierdo que inicialmente se trató como psoriasis, con mejoría parcial; sin embargo progresa con ulceración, dolor en miembro pélvico izquierdo y sangrado intermitente, por lo que acude a valoración por médico facultativo en su unidad de salud. Se realiza biopsia incisional el día 7 de enero del 2025 con reporte de carcinoma indiferenciado que infiltra dermis y tejido celular subcutáneo, ulcerado, por lo que es enviado a nuestro servicio para tratamiento especializado.
A la exploración física presenta tumor en glúteo izquierdo de 7 × 5 cm, ulcerado, móvil, doloroso a la palpación, con presencia de adenopatías inguinales móviles de 1.5 cm (Fig. 1).
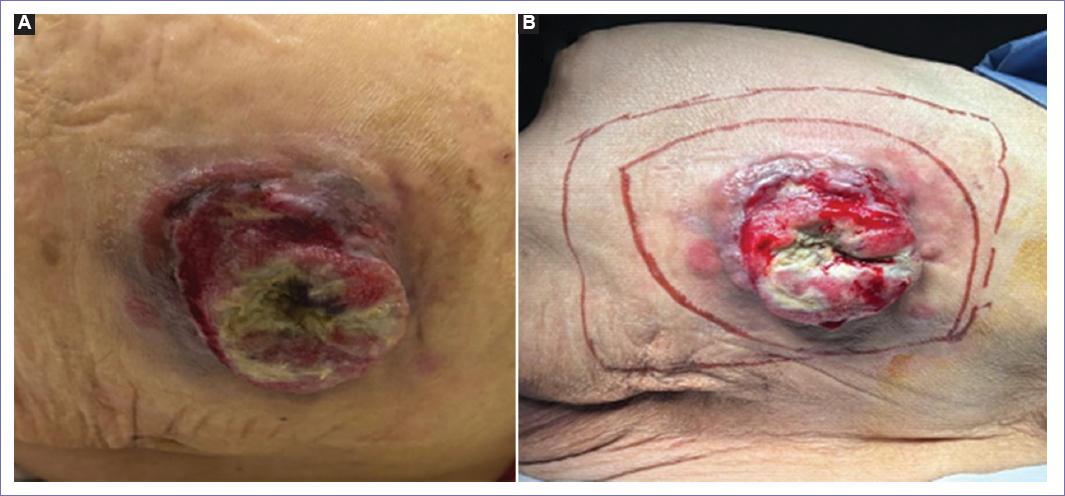
Figura 1 A: tumor en glúteo izquierdo de 7 × 5 cm, ulcerado, móvil. B: tumor en glúteo izquierdo con marcaje preoperatorio.
Se realizaron estudios de gabinete, como tomografía de abdomen (Fig. 2), donde se reporta lesión ocupante en región glútea izquierda con afectación de piel y tejido celular subcutáneo sin afectar hasta plano fascial.
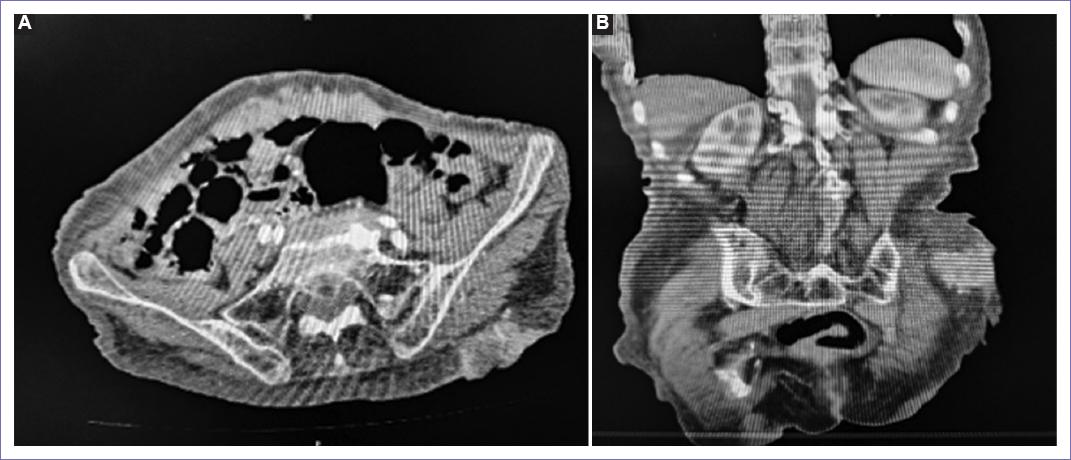
Figura 2 A: tomografía axial de abdomen en fase contrastada con presencia de tumor en glúteo izquierdo. B: imágenes coronales por tomografía abdominal contrastada donde se evidencia tumor en glúteo izquierdo hasta fascia muscular.
Se realiza revisión de laminillas, ya que la biopsia tomada por medio privado no era concluyente, reportando lesión dérmica en glúteo izquierda con marcadores de inmunohistoquímica positivos para CCM (CK20 +, CKC +, cromogranina + y sinaptofisina +, S-100 –).
Al contar adecuadamente con marcadores de inmunohistoquímica, se puede establecer de forma adecuada el diagnóstico. Se realizó resección amplia con colgajo glúteo más linfadenectomía pélvica izquierda sin complicaciones.
Se envía al servicio de patología un tumor de 12 × 11 × 4 cm fungiforme, ulcerado, así como múltiples ganglios en región inguinal de 1.5 cm y el mayor de 5 cm.
En el informe de patología se reporta CCM, de 12.5 × 10 × 3 cm con piel rugosa, ulcerada y ahulada que contiene lesión central nodular indurada y umbilicada que mide 8 × 7 × 2.5 cm, con tasa mitótica de 61 mitosis por mm2, con márgenes negativos a neoplasia. En pieza enviada como linfadenectomía pélvica se disecaron 23 ganglios, los cuales fueron positivos para malignidad (Fig. 3).
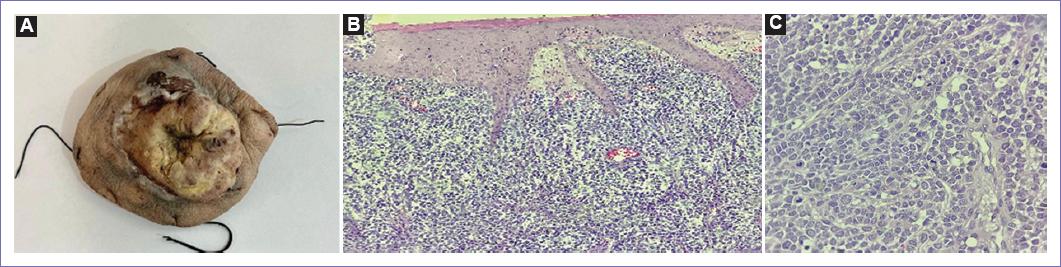
Figura 3 A: huso de piel de 12.5 × 10 × 3 cm con piel rugosa, ulcerada y ahulada que contiene lesión central nodular indurada y umbilicada que mide 8 × 7 × 2.5 cm. B y C: corte histológico donde se aprecia lesión difusa de bordes mal delimitados en dermis papilar y reticular. A mayor aumento conformado por células pequeñas, redondas y azules de aspecto epitelioide con nucleolos incospicuos con mitosis dispersas.
Respecto a nuestra paciente, se clasificó como estadio clínico IIIB (T3NIbM0), tuvo una buena evolución y no hubo ninguna barrera social para establecer el diagnóstico. Nuestro caso se envió a radiooncología para tratamiento especializado para aplicación de radioterapia 60 Gy (30 fracciones) por N+, al momento sin presentar efectos adversos al tratamiento. En la consulta de seguimiento la paciente se encuentra asintomática, con adecuada evolución y con satisfacción del adecuado manejo establecido.
Discusión
El CCM es una causa importante de morbimortalidad, puede crecer rápidamente y hacer metástasis de forma temprana; el 63% de las lesiones primarias ha crecido rápidamente en los 3 meses previos al diagnóstico9,10.
Grandes metaanálisis, como mencionan Clarke y Robbins, han demostrado que al menos la mitad de los pacientes con CCM desarrollan metástasis en los ganglios linfáticos y casi un tercio desarrollan metástasis a distancia11,12.
Estudios como el análisis de Gauci documentan el desarrollo de recurrencia locorregional en hasta la mitad de todos los casos de CCM13,14.
Se observa afectación ganglionar regional en el 45 al 91%15, como observamos en nuestro caso, en el que clínicamente se identificaron ganglios en región inguinal ipsilateral al tumor.
El tratamiento en este tipo de escenarios es realizar resección amplia, con márgenes adecuados de 1 a 2 cm, así como radioterapia16,17.
Las posibles causas asociadas a esta patología es la agresividad de la misma histología y la rareza, así como un inadecuado diagnóstico18, ya que de forma errónea se puede confundir con otros tumores, por lo que es fundamental contar con los marcadores de inmunohistoquímica, para ofrecer una intervención temprana y oportuna, para tener un control de la enfermedad, así como un periodo libre de recurrencia mayor19,20.
Al reportar nuestro caso, reforzamos la importancia de entender que el comportamiento del CCM es desalentador, ya que al momento del diagnóstico en la mayoría de los pacientes encontramos etapas localmente avanzadas, puesto que al tratarse de un CCM tras la escisión quirúrgica primaria se presenta recurrencia local en el 27-60% de los pacientes y metástasis a distancia en el 18-52%. Las tasas de supervivencia a 5 años oscilan entre el 41 y el 77%, por lo que es una entidad de pronóstico pernicioso.

