












 (pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Recientemente se ha actualizado la clasificación de las vasculitis asociadas a anticuerpos anticitoplasma de neutrófilos (ANCA), dividiéndolas en 3 grupos: a) la granulomatosis con poliangeítis (GPA), anteriormente conocida como granulomatosis de Wegener; b) la poliangeítis microscópica (PAM); y c) la granulomatosis eosinofílica con poliangeítis (o de Churg-Strauss)1. En dicha clasificación, la GPA, constituye una vasculitis necrosante de pequeños vasos, que se diagnostica con base en la afectación de las vías respiratorias superiores, de senos paranasales, faringe o de oídos, con alteraciones radiológicas pulmonares y datos de glomerulonefritis, pero, sobre todo, por la presencia de ANCA del tipo anti proteinasa-3 (cANCA-PR-3). Esta combinación de signos ha demostrado suficiente sensibilidad y especificidad en el diagnóstico de la GPA, comparable al diagnóstico histopatológico2,3.
La nefropatía en la GPA es frecuente, y se asocia a focos de necrosis focal, vasculitis y granulomas, o a nefritis túbulo-intersticial4, manifestadas clínicamente como síndrome nefrítico, hipertensión arterial, hematuria y aumento de la creatinina sérica. En algunos casos se presenta como una glomerulonefritis proliferativa extracapilar (crescéntica) con lesión renal rápidamente progresiva. En cambio, el síndrome nefrótico (SN) asociado a GPA es sumamente raro, con pocos registros anecdóticos5,6. Por otro lado, la GPA en el pulmón suele provocar vasculitis y focos de necrosis granulomatosa, manifestadas radiológicamente como lesiones nodulares y cavitadas, o en “vidrio despulido” 7. La GPA tiene una incidencia mundial de entre 10 y 20 casos por 1’000,000 de personas al año. Casi todos los pacientes presentan un curso subagudo relativamente benigno. Sin embargo, en casos aislados, la enfermedad evoluciona súbitamente a lesiones renales y pulmonares fatales de origen incierto3,8.
Aquí presentamos un caso peculiar de GPA fatal con desarrollo rápido de SN, insuficiencia renal y lesiones pulmonares difusas en una mujer joven que vive con diabetes mellitus (DM).
PRESENTACIÓN DE CASO
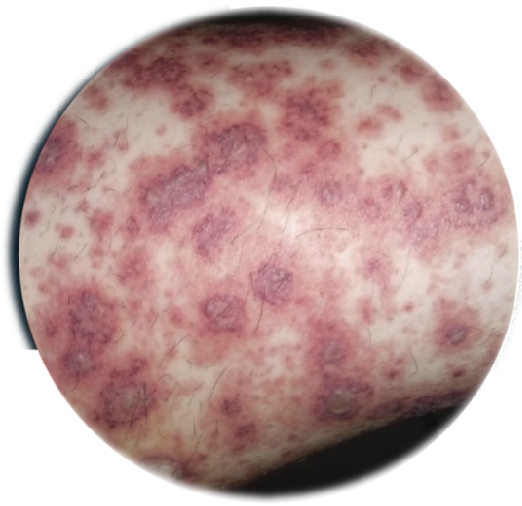
Paciente del sexo femenino, de 49 años de edad, que ingresó al servicio de urgencias del Hospital Regional Universitario de Colima, Colima, México, el 27 de septiembre de 2022 debido a edema ascendente de miembros inferiores hasta abdomen, acompañado de dolor articular en región lumbar, rodillas y muñecas, todo ello con 15 días de evolución. Dos días antes de su ingreso, notó la aparición de lesiones máculo-papulares en ambos muslos (figura 1), acompañadas de prurito y dolor quemante difuso, así como debilidad progresiva que le impidió deambular. Como antecedentes refirió ser diabética, manejada con metformina oral. Refirió haber tenido 2 partos vaginales eutócicos, el último hace 13 años.
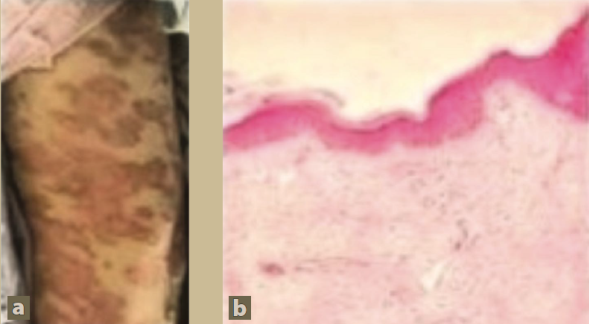
Fotos: Espinoza-Gomez et al.
Figura 1 Lesiones cutáneas en muslos y biopsia de piel (hematoxilina-eosina)
A su ingreso, la paciente se encontró pálida, postrada en cama sin poderse incorporarse, con presión arterial de 120/70 mmHg, frecuencia cardíaca de 90 latidos por minuto, temperatura de 36.6 °C y un peso de 92 kg, con índice de masa corporal de 30%. Presentaba anasarca, con predominio del edema en miembros inferiores. El examen de fondo de ojo no mostró datos de retinopatía diabética ni otras lesiones. La exploración orofaríngea reveló úlceras blanquecinas dolorosas en paladar duro. Cuello y tórax no presentaban alteraciones de relieve. Se encontró discreta ascitis y dolor a la palpación profunda en epigastrio. En ambos muslos se observaron placas eritematosas e hiperqueratósicas, de distribución difusa y bordes bien definidos. En las extremidades se halló reducción de la fuerza muscular (Daniel’s 3/5) y dolor a la movilización articular, sin datos de flogosis ni otras lesiones dermatológicas. Los pulsos estaban presentes y eran de buena intensidad, con una saturación de O2 de 94%.
Los exámenes de laboratorio al ingreso mostraron: hemoglobina 11 g/L, hematocrito 31%, plaquetas 360,000/mm3, tiempo de protrombina 14 segundos, con INR 1.2, y tiempo parcial de tromboplastina 46.6 segundos. Los electrolitos séricos reportaron: Na 132 mEq/L, K 5.2 mEq/L; urea 53 mg/dL, creatinina sérica 1.1 mg/dL, nitrógeno ureico 25 mg/dL; glucemia casual 173 mg/dL; albúmina sérica 2.1 g/dL; AST 18 UI/L, ALT 20 UI/L, DHL 270 UI/L, fosfatasa alcalina 270 UI/L, bilirrubinas totales 1.2 mg/dL; colesterol total 174 mg/dL y triglicéridos 214 mg/dL. El examen general de orina mostró proteinuria de 600 mg/dL, eritrocitos 100 por campo. La cuantificación de proteínas en orina de 24 horas fue de 6.8 g en 520 mL. La gasometría arterial reveló: pO₂ 85 mmHg, pCO₂ 32 mmHg, HCO₃ 18 mEq/L y saturación de O2 de 96%.
Una tomografía axial computarizada de tórax realizada el día del ingreso mostró derrame pleural bilateral e imágenes sugestivas de “vidrio despulido” en la base pulmonar derecha (figura 2a). Un ultrasonido abdominal evidenció riñones aumentados de tamaño (12 y 13 cm, respectivamente), con disminución general de la densidad y pérdida de la relación corteza-médula.
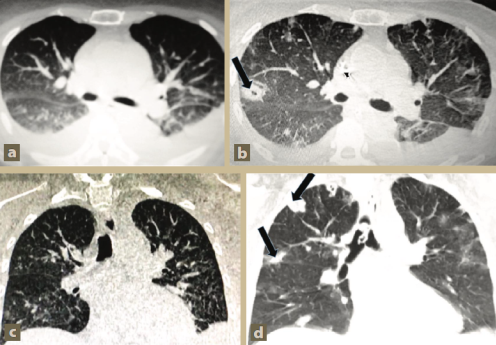
Fotos: Espinoza-Gomez et al.
Figura 2 Tomografía axial computarizada de tórax al ingreso y después de 15 días de hospitalización
Con base en los hallazgos de proteinuria significativa, anasarca e hipoalbuminemia, se diagnosticó SN asociado a la DM. Se inició tratamiento con furosemida intravenosa (20 mg cada 8 horas) e insulina de acción rápida para mantener glucemias por debajo de 120 mg/dL.
Durante las siguientes 48 horas, la paciente presentó oliguria (<400 mL en 24 h), con incremento en la creatinina sérica (de 1.1 a 2.3 mg/dL) y de potasio (hasta 6.3 mEq/L). La hemoglobina descendió de 10 a 7.5 g/dL. El recuento de reticulocitos fue de 2%, la velocidad de sedimentación globular de 24 mm/h, y la proteína C reactiva fue positiva (17.4 mg/dL).
En los días posteriores desarrolló disnea y polipnea con saturación de O2 <80%, lo cual obligó al inicio de hemodiálisis 3 veces por semana a través de catéter subclavio, así como respiración asistida con mascarilla no invasiva.
Durante los siguientes 3 días, presentó mejoría clínica: disminuyeron la disnea, el edema, las lesiones dérmicas y el estado general. Sin embargo, en el undécimo día, desarrolló súbitamente disnea progresiva con hipoxemia (60 mmHg), frecuencia cardíaca de 109 lpm e hipotensión arterial progresiva. Se diagnosticó probable choque séptico y síndrome de dificultad respiratoria aguda (SDRA). Se inició tratamiento con meropenem intravenoso (1 g cada 12 horas), norepinefrina (45 µg/minuto en infusión continua por catéter venoso central) y heparina, ante la sospecha de tromboembolia pulmonar, respaldada por la elevación del dímero D (700 ng/mL). Las sesiones de diálisis continuaron, aunque con bajo flujo debido a la hipotensión persistente.
Una nueva TAC de tórax, realizada 15 días después de la inicial, mostró la aparición rápida de lesiones nodulares difusas bilaterales, algunas de ellas cavitadas (figura 2b).
Se realizaron cultivos de sangre, esputo y orina, resultando todos negativos, al igual que los niveles de procalcitonina sérica (0.33 ng/dL) y las baciloscopias de esputo. Los anticuerpos antinucleares, el factor reumatoide, y las pruebas para VIH, hepatitis B y C fueron negativos. Sin embargo, los anticuerpos contra proteinasa 3 (cANCA-PR3) resultaron positivos (150 UR/mL por ELISA); mientras que los anticuerpos antimieloperoxidasa (pANCA-MPO) y anti-membrana basal fueron negativos.
Durante los días siguientes, el estado de la paciente continuó deteriorándose, con mayor esfuerzo respiratorio, hipoxemia y acidosis metabólica (PO₂/FiO₂: 160 mmHg; pH: 7.1; HCO₃: 10.7 mEq/L), por lo que se inició ventilación mecánica invasiva. En el día 17 de hospitalización, la paciente presentó un paro cardíaco que no respondió a maniobras de reanimación.
Los familiares rehusaron inicialmente la realización de una biopsia renal y finalmente también la autopsia, aunque firmaron carta de consentimiento para la publicación del caso, respetando la confidencialidad y conforme a las normas vigentes para el manejo de pacientes en instituciones públicas en México.
DISCUSIÓN
Si bien no contamos con estudios histopatológicos de riñón ni pulmón, según las nuevas guías de la EULAR y el ACR para el diagnóstico de vasculitis asociadas a ANCA, el caso reúne los elementos necesarios para establecer el diagnóstico, especialmente considerando que los cANCA-PR3 son altamente específicos2.
En estos pacientes el diagnóstico diferencial debe incluir vasculitis con nefritis y neumonitis sin ANCA, como:
Lupus eritematoso generalizado (LEG).
Púrpura de Henoch-Schönlein.
Poliarteritis nodosa.
Síndrome de Goodpasture.
Vasculitis leucocitoclástica de Churg-Strauss (asociada a pANCA-MPO y eosinofilia).
Nefritis secundaria a sepsis.
Todas estas condiciones se descartaron según las guías EULAR/ACR2,4.
En nuestra paciente predominó el SN, por lo que el diagnóstico inicial fue nefropatía diabética, que progresó a trombosis de venas renales y lesión renal aguda (LRA), lo que originó la oligoanuria, dolor abdominal y edema pulmonar agudo. Sin embargo, la presencia de úlceras orales, de nódulos cavitados en pulmón, la elevación de VSG y PCR, así como la presencia de cANCA-PR-3 nos condujeron al diagnóstico de granulomatosis con poliangitis (GPA), con deterioro renal y respiratorio súbito, a pesar del tratamiento con metilprednisolona.
La presencia de SN en la GPA asociada a DM es poco común6 y el mecanismo de la proteinuria masiva en estos pacientes es incierto, aunque algunos autores han atribuido este fenómeno a daño túbulo intersticial secundario, más desnutrición y aumento de la permeabilidad capilar glomerular4,9. Como sea, esta proteinuria masiva en pacientes con nefropatía por ANCA aumenta la insuficiencia renal crónica y la mortalidad10.
Por su parte, el rápido desarrollo de lesiones pulmonares con SDRA, parece ser igualmente un fenómeno poco frecuente en la GPA7,8. En nuestro caso inicialmente consideramos las siguientes posibilidades:
Edema pulmonar agudo secundario a la falla renal.
Una neumonía infecciosa atípica.
Una trombosis pulmonar.
Sin embargo, el tipo de lesiones radiológicas, la ausencia de cultivos positivos, y los niveles relativamente bajos de dímero D, o de procalcitonina C, así como la presencia de cANCA-PR3, nos hace suponer más bien que todo se debió a vasculitis pulmonar por GPA con hemorragia pulmonar intensa, dada la súbita reducción de función respiratoria y de hemoglobina con datos de shock, aunque no hayamos observado sangrado por vía orotraqueal.
Respecto a las lesiones dérmicas, aunque no se observaron cambios histológicos propios de vasculitis (figura 1), nos parece claro que este fenómeno formó parte de la propia vasculitis sistémica, semejante a lesiones similares ya descritas11.
El tratamiento de la GPA incluye el uso de corticoides en bolo intravenoso, lo cual no se intentó en este caso debido a la presencia de DM y a la incertidumbre sobre el tipo de lesión renal. En refractarios, se ha utilizado ciclosporina o de anticuerpos monoclonales como el rituximab, aunque con resultados poco satisfactorios11. Resulta inquietante considerar que un diagnóstico más temprano de esta patología podría haber cambiado su pronóstico, al haber iniciado inmunosupresores o plasmaféresis en forma oportuna, tal como se ha informado en casos de GPA con daño pulmonar severo8,11. Esto no fue posible, debido a que los estudios inmunológicos no son de fácil acceso en hospitales generales públicos de México y a la negativa para obtener biopsia renal.
CONCLUSIÓN
En conclusión, la GPA representa un importante reto diagnóstico ante el cual los médicos en hospitales generales debemos mantener un alto índice de sospecha, no solamente cuando existen lesiones típicas como sinusitis, afección pulmonar significativa o nefritis aguda, sino también ante manifestaciones atípicas como el SN y el desarrollo de SDRA en ausencia de procesos infecciosos o congestivos manifiestos. Se recomienda incluir el estudio de cANCA en el protocolo diagnóstico de todo paciente con SN, hematuria e insuficiencia renal, independientemente de la realización de biopsia renal.

