











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkPresentación del caso
Presentamos el caso de una familia con una historia de trastornos de la conducción cardiaca y miocardiopatía asociada en la que se identificó una mutación no descrita en el gen TNNI3K.
El caso índice era un varón de 44 años, sin antecedes de interés, que ingresó por un síncope de perfil cardiogénico. En el electrocardiograma (ECG) se observó un bloqueo de rama izquierda, por lo que se realizó un estudio electrofisiológico que mostró un trastorno de la conducción intrahisiano con un AH de 100 ms y un HV de 61 ms. Se decidió el implante de un marcapasos bicameral. Presentaba el ventrículo izquierdo no dilatado con función sistólica (fracción de eyección del ventrículo izquierdo [FEVI]) levemente reducida (48%), que se interpretó como probablemente secundaria a la asincronía intraventricular. No se observaron valvulopatías funcionalmente relevantes ni datos de hipertensión pulmonar significativa. El estudio etiológico se completó con una resonancia magnética (RM) cardiaca realizada a los 3 meses, que mostró un ventrículo izquierdo de dimensiones en el límite superior de la normalidad (volumen telediastólico 99 ml/m2) con FEVI del 43% y un foco de edema inferolateral con fibrosis subendocárdica del 60% en dicho segmento (Fig. 1A y B). Una tomografía computarizada coronaria descartó la presencia de estenosis significativas. Se inició tratamiento neurohormonal, a pesar de lo cual al año desarrolló disfunción grave del ventrículo izquierdo, persistiendo asintomático. En la interrogación del dispositivo, el porcentaje de estimulación era inferior al 10% y se objetivaron varias salvas de taquicardia ventricular no sostenida de hasta ocho complejos, por lo que ante la presencia de disfunción ventricular grave y fibrosis se decidió realizar un up-grade a un desfribrilador automático implantable - terapia de resincronización cardíaca (DAI-TRC), con posterior recuperación completa de la FEVI.
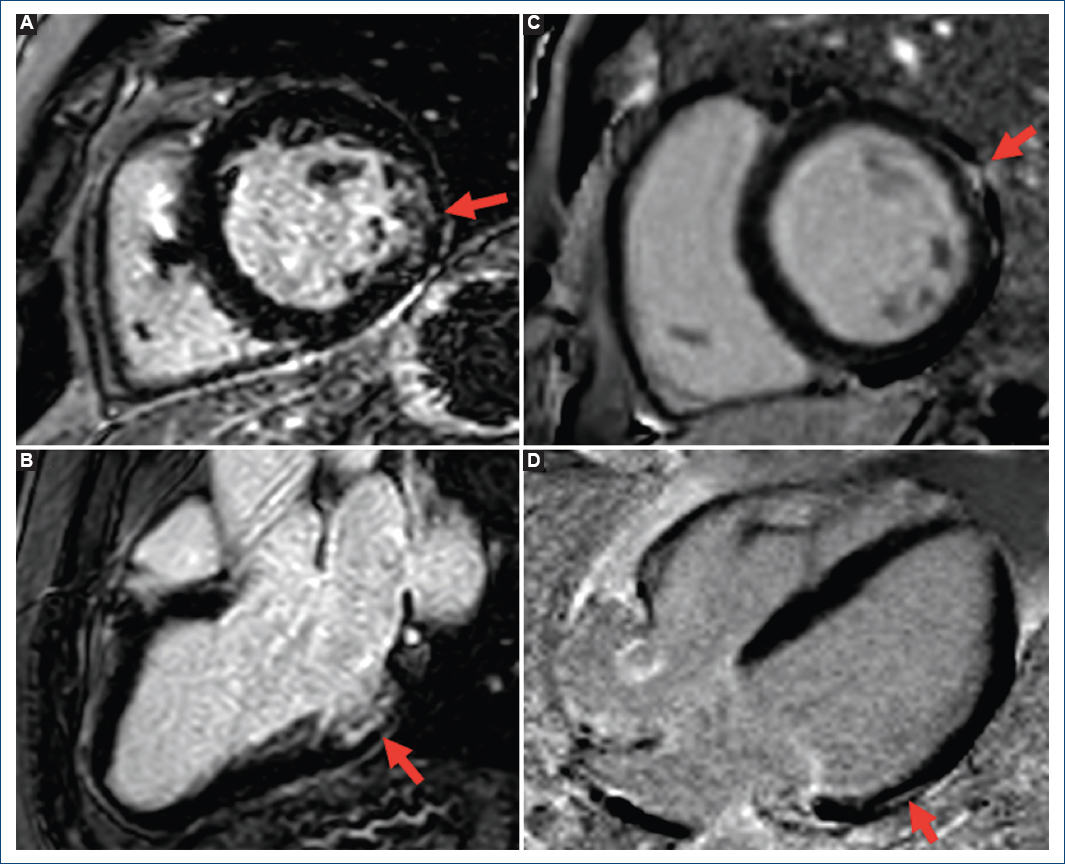
Figura 1 A y B: resonancia magnética (RM) del caso índice, secuencias de realce tardío de gadolinio (RTG), eje corto y eje largo, que muestra fibrosis subendocárdica del 60% inferolateral. C y D: resonancia magnética de la madre del caso índice, secuencias de realce tardío de gadolinio, eje corto y eje largo, que muestra fibrosis lineal intramiocárdica lateral basal.
En la evaluación inicial se elaboró un árbol familiar y se realizó un cribado en los familiares de primer grado (Fig. 2). Su madre había sido estudiada a los 60 años por síncopes de perfil cardiogénico con hallazgo de bloqueo de rama izquierda, objetivándose posteriormente en un estudio electrofisiológico un trastorno de la conducción infrahisiano y se le implantó un marcapasos. Desde el punto de vista estructural presentaba un ventrículo izquierdo no dilatado con FEVI del 50% y en la RM realizada 2 años después del implante del marcapasos se observó una FEVI del 46% con acinesia limitada al segmento medio lateral, sin observarse fibrosis. Por otro lado, su hermano había sido estudiado previamente por extrasistolia ventricular de baja densidad sintomática con un bloqueo de rama derecha en el ECG. Estructuralmente presentaba un ventrículo izquierdo ligeramente dilatado con FEVI normal y un foco de fibrosis lineal intramiocárdica lateral basal en la RM (Fig. 1 C y D). El abuelo materno era portador también de marcapasos desde los 52 años por un bloqueo auriculoventricular completo y había fallecido en la octava década de la vida por insuficiencia cardíaca.
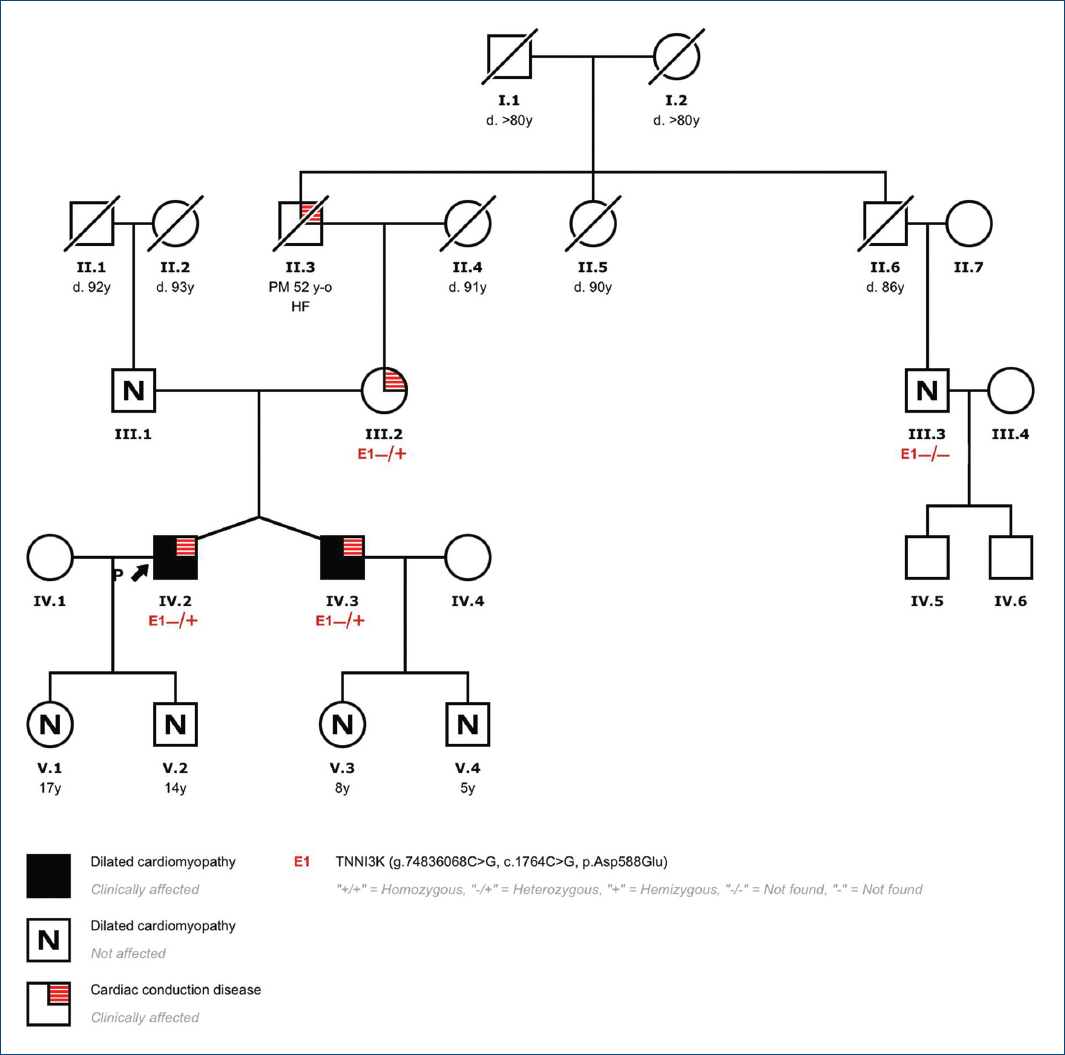
Debido a la historia familiar, se pensó en una etiología hereditaria y se realizó un estudio genético amplio (251 genes cardiovasculares asociados a miocardiopatías y arritmias cardíacas). Se identificó la variante de significado incierto p.Asp588Glu en el gen TNNI3K en heterocigosis. Este es un gen candidato (de evidencia limitada) para el desarrollo de miocardiopatía dilatada y trastornos de la conducción cardiaca. Codifica la cinasa de interacción con TNNI3, una proteína formada por 835 aminoácidos, localizada en el cromosoma 1 humano. En ratones ha demostrado desempeñar un papel importante en la diferenciación y la contractilidad cardíacas. Su sobreexpresión acelera la progresión de la miocardiopatía en ratones y aumenta la duración del intervalo PR1,2.
Son pocas las variantes causales descritas en este gen, siendo todas ellas de tipo missense, aunque los datos que las asocian al desarrollo de este fenotipo solapado de trastornos de la conducción, arritmias supraventriculares y miocardiopatía dilatada parecen robustos y concluyentes. Dos de estas variantes se localizan cercanas a la que se identificó en esta familia. La variante p.Gly526Asp se describió en 2014 en una gran familia con ocho portadores con un fenotipo solapado de arritmias supraventriculares, miocardiopatía dilatada y trastornos de la conducción, y tres no portadores no afectos (en edad infantil). Aunque no se describían resonancias cardiacas, se halló fibrosis intersticial en una biopsia endomiocárdica del caso índice3. La variante p.Thr539Ala fue descrita un año más tarde en cuatro miembros de una familia con trastornos de la conducción auriculoventricular y episodios de taquicardia ectópica de la unión4. Recientemente se han descrito otras dos variantes (p.Ile512Thr y p.His592Tyr) asociadas a miocardiopatía dilatada, trastornos de la conducción, muerte súbita y arritmias supraventriculares. En este trabajo se realizaron estudios in vitro en los que se comprobaron niveles elevados de autofosforilación de TNNIK3, indicando una función de la cinasa aumentada, no presentes en los controles5.
Estas variantes, al igual que la identificada en nuestro paciente, se encuentran en el dominio cinasa (aminoácidos 463-723) de la enzima, que podría corresponder a un hot-spot. En 2019 se describió otra variante missense (p.Glu768Lys) en 23 portadores de tres familias multigeneracionales con un fenotipo de arritmias supraventriculares que se solapaban con miocardiopatía dilatada y trastornos de la conducción, localizada muy cercana a este dominio6.
En nuestro caso se evaluó la cosegregación familiar de la variante, siendo su madre y su hermano mellizo portadores de la variante en TNNI3K. Se amplió el estudio de cosegregación con estudio de un primo materno, que resultó no portador, con ECG y estudio estructural normales. Con estos resultados, la variante fue recategorizada a posiblemente patogénica, según los criterios del American College of Genetics and Genomics (ACMG) y las adaptaciones de ClinGen que aplican a este caso (PM2, PP3, PM1_sup, PP1_mod, PP4).
Presentamos, por tanto, la sexta variante missense en TNNI3K de la literatura científica, que cosegrega en una familia con un fenotipo solapado de miocardiopatía dilatada y trastornos de la conducción, describiendo por primera vez la presencia de realce tardío de gadolinio como hallazgo asociado.

