











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La discinesia ciliar primaria (DCP) es un trastorno genético heterogéneo autosómico recesivo caracterizado por disfunción de la motilidad ciliar que ocasiona enfermedades de las vías respiratorias superiores e inferiores, defectos de lateralidad de los órganos y dificultades reproductivas1.
La confirmación diagnóstica de la DCP es todo un desafío porque se basa en la presencia de defectos ultraestructurales del axonema ciliar mediante microscopía electrónica de transmisión (MET), la medición del óxido nítrico nasal (nNO), el análisis del patrón y la frecuencia del batido ciliar mediante videomicroscopía de alta velocidad (VMAV), la inmunofluorescencia de las proteínas ciliares (IF) y las pruebas genéticas2,3.
Actualmente existen más de 50 genes implicados en esta enfermedad reconocidos por su alteración en la estructura ciliar1; además, se han establecido las relaciones genotipo-fenotipo que caracterizan mejor algunas mutaciones genéticas y las manifestaciones clínicas en la población con DCP4.
Por el avance de los estudios genéticos se han encontrado variantes de significado incierto (VUS, variants of uncertain significance) en pacientes con un fenotipo clínico compatible con DCP5, y en pacientes que no presentan la enfermedad, por lo que es importante la correcta interpretación de los resultados genéticos y no solo basarse en ellos para hacer el diagnóstico de DCP4.
El objetivo de este reporte es describir una VUS en el gen DNAI1 en un paciente con fenotipo clínico y MET compatible con DCP.
Caso clínico
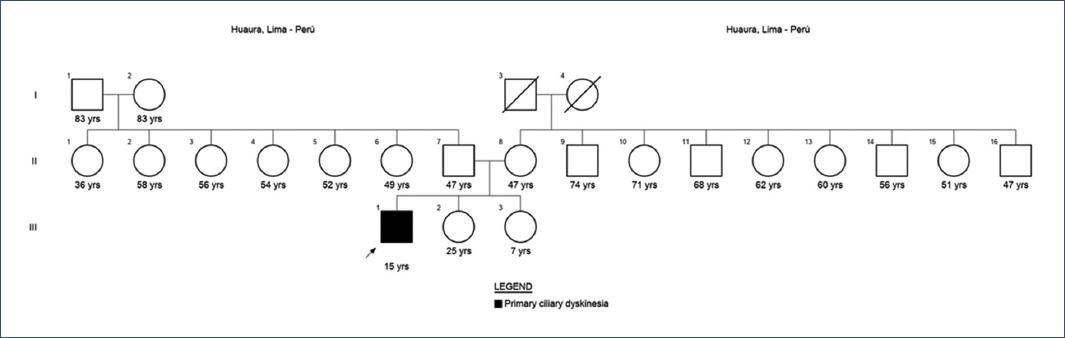
Se presenta el caso de un paciente de sexo masculino de 15 años que acudió a consulta por tos húmeda crónica con expectoración verdosa, disnea con moderados esfuerzos (escala de disnea modificada del British Medical Research Council grado 2) y disminución de la actividad física. El paciente nació a término por parto eutócico con 39 semanas (segunda gestación), peso al nacer 3300 g y APGAR 81-95, sin complicaciones durante el parto. A las horas de nacido presentó dificultad respiratoria y fue diagnosticado de neumonía neonatal, requirió ventilación mecánica por 8 días y fue dado de alta a los 15 días de vida. Sus padres no tienen consanguinidad ni antecedentes familiares de patologías respiratorias (Fig. 1).
Al año de edad, el paciente fue hospitalizado por neumonía y a partir de los 2 años de edad presentó 4 o 5 episodios por año de tos húmeda, sibilancias y dificultad respiratoria, y fue manejado como síndrome obstructivo bronquial recurrente más infecciones bacterianas del tracto respiratorio inferior. Durante estos episodios recibió tratamiento con salbutamol, budesonida y antibióticos, con mejoría parcial. También presentó 3 o 4 episodios al año de otitis media no supurada y recibió antibioticoterapia en cada episodio.
En la exploración física, el paciente estaba lúcido, no tenía fiebre y sus parámetros vitales estaban dentro de los valores normales. Presentaba pectus excavatum y en la auscultación pulmonar había crépitos difusos bibasales de predominio derecho. El resto de la exploración no mostró alteraciones. El análisis del estado nutricional reveló delgadez grave (peso 38 kg, talla 155 cm, IMC/E –3.32 z, T/E –1.96 z).
En los exámenes auxiliares, la prueba de tuberculina fue de 0 mm, y en el hemograma no se evidenciaron leucocitos ni neutrofilia. Por el historial de tos húmeda crónica, se plantean como diagnósticos diferenciales fibrosis quística, inmunodeficiencia primaria, asma no controlada, bronquitis bacteriana prolongada, rinosinusitis y enfermedad por reflujo gastroesofágico, entre otros. Se realizó dosaje de inmunoglobulinas séricas IgM, IgG, IgA, subclases de IgG e IgE, que resultaron normales. La citometría de flujo con recuento y fenotipo de linfocitos T, linfocitos B y linfocitos NK estaba en rangos normales para la edad. La prueba de cloro en sudor por iontoforesis con pilocarpina fue de 20 mmol/l (negativo). La ecocardiografía fue normal. La otoscopia y la timpanometría también fueron normales.
Los estudios radiológicos mostraron opacidad paracardiaca derecha que borraba parcialmente el borde cardiaco derecho y refuerzo de la trama broncovascular bilateral (Fig. 2A), alteración de los senos paranasales (Fig. 2B), bronquiectasias quísticas del lóbulo medio (Fig. 2C) y bronquiectasias cilíndricas en ambos lóbulos inferiores.
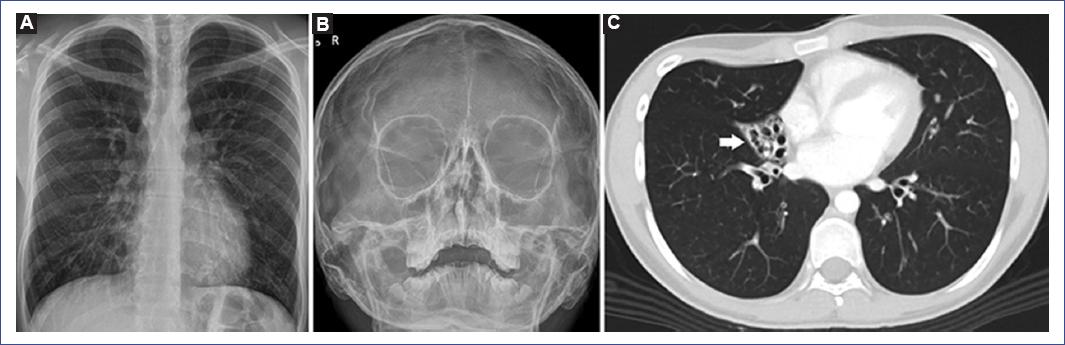
Figura 2 A: radiografía de tórax. Se observan opacidad paracardiaca derecha que borra parcialmente el borde cardiaco derecho y opacidades hiliobasales bilaterales. Además, refuerzo de la trama broncovascular bilateral. B: radiografía de senos paranasales (incidencia Waters). Se evidencia engrosamiento mucoso periférico en ambos senos maxilares con predominio izquierdo, opacificación del seno frontal izquierdo parcialmente desarrollado, seno frontal derecho aún no desarrollado y opacificación de celdillas etmoidales. C: tomografía computarizada de tórax con contraste. En corte axial en ventana de parénquima se observan bronquiectasias quísticas en el lóbulo medio (flecha).
Se realizaron broncoscopía y lavado broncoalveolar, con aislamiento de Streptococcus pneumoniae sensible a la clindamicina, la eritromicina, el cloranfenicol y la vancomicina, y resistente a la oxacilina y a la trimetoprima-sulfametoxazol. Recibió cobertura antibiótica con ceftriaxona por 14 días, con buena evolución. Por ser el reflujo gastroesofágico una de las causas de tos húmeda crónica, se realizó endoscopia digestiva alta que reportó hernia hiatal tipo 1 y gastritis superficial leve, por lo que recibió tratamiento con omeprazol y mosaprida. También se realizó una espirometría, que reportó VEF1/CVF de 67.9, menor que el límite inferior normal (LIN: 87.5), con VEF1 del 67% p, CVF del 86% p y posbroncodilatador con VEF1/CVF de 69.8, menor que el límite inferior normal (LIN: 87.5), VEF1 67% p, CVF 84% p; patrón obstructivo moderado, sin reversibilidad.
Al aplicar la puntuación PICADAR6 se obtuvieron 8 puntos: nacido a término (2), síntomas respiratorios en la etapa neonatal (2), ingreso a la unidad de cuidados intensivos neonatales (2), rinitis persistente (1) y síntomas crónicos del oído (1). El estudio de la ultraestructura ciliar por MET informó que, en los cortes transversales de los cilios, la mayor parte presentaban la distribución clásica de 9 + 2 pares de microtúbulos periféricos que rodean un par central, pero algunos de los microtúbulos presentaban distribución de 6 + 0, 7 + 1, 8 + 0, 8 + 3, 8 + 1, 9 + 0 y 9 + 1, fusión de los microtúbulos periféricos, ausencia de pares de microtúbulos periféricos y en su lugar microtúbulos individuales, y otros con ausencia del par central envueltos por la membrana ciliar (Figs. 3A y C). Además, tomando en cuenta los criterios cuantitativos de Carlén y Stenram7, se observó que casi la totalidad de los microtúbulos presentaban ausencia de los brazos internos de dineína (IDA), los que promedian 0.75 (valores referenciales: 3.0-5.0), y la mayoría de ellos mostraban ausencia de los brazos externos de dineína (ODA), con promedio de 1.7 (valores referenciales: 7.5-9.0) (Fig. 3B). Estos hallazgos fueron sugerentes de DCP.
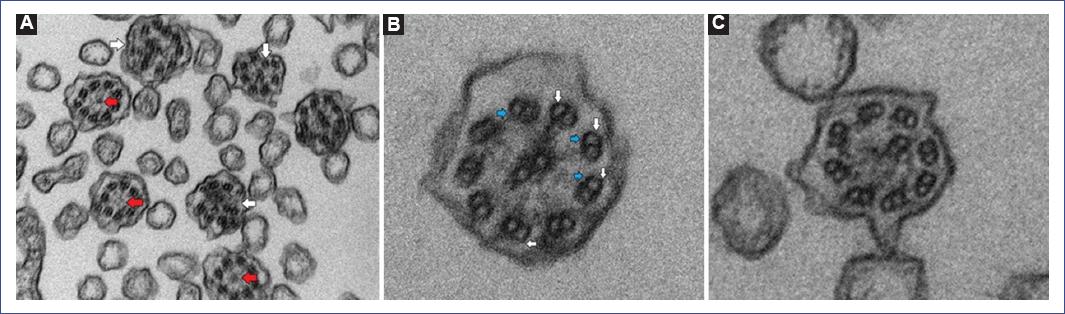
Figura 3 Microscopía electrónica de transmisión. A: en varias secciones transversales ciliares se observan desorganización microtubular (flechas blancas) y ausencia del par de microtúbulos centrales 9 + 0 (flechas rojas) (aumento × 60.000). B: corte transversal de cilio respiratorio en el que se observa que la mayor parte de los microtúbulos periféricos muestran ausencia de los brazos internos (flechas azul celeste) y externos (flechas blancas) de dineína (aumento × 85.000). C: corte transversal de cilio respiratorio en el que se observa una organización microtubular alterada 8 + 2, con ausencia de uno de los microtúbulos periféricos y desplazamiento del par central. La mayor parte de los microtúbulos periféricos muestran ausencia de los brazos internos y externos de dineína (aumento × 85.000).
Se realizó un panel genético para inmunodeficiencias (Veritas®) mediante secuenciación de ADN de nueva generación y se encontró una variante homocigota en el gen DNAI1 (NM_012144.4): c.1489+5G>A. Esta variante se encuentra ausente en cohortes poblacionales como gnomAD (PM2) y es homocigota, lo que indica que se encuentra en trans (PM3). Es una variante que afecta a la región de empalme con predicción de ser deletérea en herramientas in silico (dbscSNV 1) (PP3). Clasificamos la variante como VUS según las recomendaciones establecidas por el American College of Medical Genetics and Genomics y la Association for Molecular Pathology8. A pesar de ser una VUS, debido a los hallazgos clínicos y de la MET, el diagnóstico del paciente fue DCP.
El paciente evolucionó satisfactoriamente luego de la terapia con nebulizaciones (salbutamol al 0.5% y solución salina hipertónica al 3%), azitromicina 400 mg por vía oral tres veces por semana, N-acetilcisteína 400 mg por vía oral cada 8 horas y fisioterapia respiratoria, sin reporte de reacciones adversas. Durante 1 año de seguimiento, el paciente tuvo una exacerbación respiratoria viral que requirió hospitalización por 5 días y luego se mantuvo estable.
Discusión
La DCP es una enfermedad genética rara que a menudo está subdiagnosticada debido a falta de conocimiento y acceso limitado a las pruebas e instalaciones de diagnóstico1,9,10.
Los síntomas clínicos típicos y los defectos de clase 1 (ODA, ODA-IDA, desorganización microtubular-IDA) del axonema ciliar en la MET son diagnósticos de DCP, aunque en el estudio genético se identifiquen VUS11, como en este caso.
El análisis de VMAV debe ser realizado por personal experimentado como parte del trabajo de diagnóstico en pacientes con sospecha de tener DCP, lo cual puede perjudicar la disponibilidad de la prueba en muchos centros2. Los porcentajes de bordes discinéticos y el índice de inmotilidad ciliar altos podrían ser fiables para el diagnóstico de DCP3.
El nNO bajo, teniendo en cuenta que es relativamente fácil de determinar en niños mayores de 6 años y en adultos, puede establecer un diagnóstico presuntivo de DCP en pacientes con un fenotipo clínico compatible; sin embargo, en niños menores de 6 años es muy difícil de determinar con un analizador de quimioluminiscencia con la técnica del cierre del velo del paladar, y puede ser menos discriminatorio de DCP2. Los valores de nNO pueden disminuir transitoriamente en presencia de infecciones respiratorias virales agudas o sinusitis, y está indicado establecerlos en dos ocasiones separadas3.
No hay estudios que hayan podido demostrar la precisión de la IF como prueba diagnóstica de DCP porque solo puede examinar la inadecuada localización de la proteína relacionada con mutaciones genéticas2.
Los estudios de genes reportados hasta la fecha han logrado establecer relaciones genotipo-fenotipo; sin embargo, la presencia de VUS en los estudios genéticos hace necesaria la realización de estudios adicionales para corroborar su patogenicidad4.
Los genes implicados en esta enfermedad varían según la raza. Un estudio de variantes genéticas encontró que en la población latinoamericana predominan los genes DNAAF4, DNAH11, DNAH5 y DNAAF19. Al respecto, el estudio genético de nuestro caso fue una VUS en el gen DNAI1 (c.1489+5G>A), y aunque el fenotipo de nuestro paciente fue compatible, se requiere más evidencia, como estudios funcionales, para poder reclasificarla como patogénica o probablemente patogénica1.
Las mutaciones en el gen DNAI1 ocasionan alteraciones en la MET (defecto del brazo externo de dineína), el nNO (bajo) y la VMAV (mínimo movimiento), además de las características típicas como tos crónica, bronquiectasias, rinitis crónica, distrés respiratorio neonatal, defectos de lateralidad y problemas de fertilidad12. Nuestro paciente tenía un fenotipo típico de DCP (PICADAR 8 puntos)6, bronquiectasias cilíndricas basales bilaterales y del lóbulo medio (Fig. 2), y en la MET mostró alteraciones ultraestructurales compatibles con DCP (Fig. 3); esto último podría correlacionarse con la alteración del gen DNAI1 encontrada en el estudio genético.
Un estudio en un hospital del Reino Unido encontró que 16 de 101 (15.8%) pacientes tenían una o más VUS; todos los pacientes presentaban alteraciones en la MET y fenotipo típico de DCP5. En el futuro, el número de genes relacionados con la DCP probablemente aumente debido a la mayor disponibilidad de estudios genéticos; sin embargo, interpretar el resultado de estas pruebas puede ser todo un desafío por el alto número de VUS en una enfermedad con heterogeneidad de locus9, por lo cual la reclasificación de las VUS en variantes patogénicas será un reto y no se pueden asumir como causa de la enfermedad con fines de diagnóstico4.
Aproximadamente el 25% de los pacientes con DCP no son detectados por las pruebas genéticas1, por lo que un resultado negativo no descarta DCP en un paciente con fenotipo compatible. A pesar de ello, es importante el estudio genético, pues permitirá en un futuro establecer con mayor detalle la relación genotipo-fenotipo1,12, que ayudará en el pronóstico y la detección de complicaciones13, además de la posibilidad de participación en tratamientos dirigidos utilizando terapias genéticas o de transcripción4.
Las tecnologías de secuenciación de ADN de nueva generación (NGS, next-generation sequencing) en términos de secuenciación del exoma completo (WGS, whole genome sequencing) permiten la acumulación de datos de pacientes que dieron negativo para los genes conocidos de DCP, que serán útiles para una mayor investigación sobre la identificación de nuevos genes o mutaciones candidatas1.
Consideramos que la identificación oportuna y precisa de DCP en pacientes que tienen fenotipo compatible con esta enfermedad permitirá dar el tratamiento adecuado y el asesoramiento genético para esta enfermedad hereditaria multisistémica.
Nuestro reporte presenta algunas limitaciones. No realizamos estudios de nON, VMAV e IF por no estar disponibles en nuestro país. Tampoco, por cuestiones económicas y de acceso, se pudo realizar la búsqueda de la variante genética en los familiares de primer grado, ni hacer estudios funcionales que podrían ayudar a reclasificar la variante genética encontrada.
A pesar de ello, consideramos que las fortalezas del diagnóstico de DCP en nuestro caso son los hallazgos tipo 1 en la MET y el fenotipo clínico compatible con DCP; esto hace que la presencia de una VUS refuerce más el diagnóstico. El reporte brinda un aporte a la variante genética descrita, especialmente en un paciente latinoamericano, pues la mayoría de los informes y de los estudios genéticos provienen de América del Norte y de Europa9.
En conclusión, es importante el reporte de una VUS en pacientes con fenotipo clínico y hallazgos de clase 1 en la MET ciliar compatibles con DCP, con el fin de aportar a la reclasificación de variantes genéticas. Es necesario fortalecer los centros latinoamericanos de estudio de DCP y promover redes de colaboración para conocer en detalle las características fenotípicas y genéticas de estos pacientes.

