(pdf)
(pdf)




 SciELO
SciELO  SciELO
SciELO 
 Permalink
PermalinkAntecedentes
La Enfermedad de Fabry (EF) fue reconocida inicialmente con el nombre de “púrpura papulosa hemorrágica”,1 siendo los dermatólogos Johannes Fabry y William Anderson, quienes describieron por primera vez el “angioqueratoma corporis diffusum” en 1898.2 Se reconoció tempranamente como una enfermedad vascular sistémica y más tarde como un trastorno de almacenamiento de lípidos.3,4 En 1963 se identificó la acumulación de glicolípidos ceramida trihexósida (ahora llamado globotriaosilceramida (Gb3 o GL-3)) y galabiosilceramida en una variedad de diferentes tipos de células;5 varios años después, el defecto se estableció como actividad insuficiente de la enzima ceramida trihexosidasa que cataliza la escisión hidrolítica de la molécula terminal de galactosa de Gb3.6 La naturaleza ligada al cromosoma X de la enfermedad se reconoció por primera vez en 1965.7
Definición
La EF (OMIM #301500),8 o angioqueratoma corporis diffusum, es un trastorno hereditario poco común y muy debilitante del metabolismo de los glucoesfingolípidos, asociado con complicaciones renales, cardíacas y cerebrovasculares.9
La deficiencia de una hidrolasa lisosomal, agalactosidasa, conduce a la acumulación progresiva de glucoesfingolípidos (principalmente ceramida trihexósido (GL-3 o Gb3)) en la mayoría de los tejidos viscerales, incluidas las células vasculares (células endoteliales y del músculo liso), células cardíacas (cardiomiocitos y células valvulares), células renales (células tubulares y glomerulares), células nerviosas y principalmente en los lisosomas del endotelio vascular. La acumulación progresiva de glucoesfingolípidos endoteliales da lugar a isquemia e infarto tisular y conduce a las principales manifestaciones clínicas de la enfermedad.10
Epidemiología
Se ha estimado que la prevalencia de EF es de alrededor de 1 por cada 40,000 varones,11 otro estudio encontró 12 de 37,104 neonatos varones consecutivos con mutaciones específicas en el gen de la α-galactosidasa A (ligado a X Xq22.1, como se describirá en Fisiopatología);12 por su parte, Clarke estimó en 2007 1 paciente con EF por cada 55,000 varones nacidos.13
Las manifestaciones cerebrovasculares son frecuentes tanto en el grupo homocigoto como en el heterocigoto sintomático. Más importante aún, estas manifestaciones pueden ser la primera indicación de la enfermedad.10 En un estudio prospectivo de 721 pacientes de 18 a 55 años, el 4.9% de los pacientes varones y el 2.4% de las mujeres presentaban mutaciones biológicamente significativas en el gen GLA.14
En México no hay estudios de prevalencia de la EF ni de sus manifestaciones neurológicas, probablemente por su baja incidencia y por la dificultad diagnóstica que esta implicaba hasta hace algunos años. En general, la EF y su manifestación de Enfermedad vascular cerebral (EVC) queda oculta en la clasificación de etiología criptogénica, un diagnóstico final frecuente de la EVC. En relación a esto cabe mencionar el estudio de Barinagarrementeria y cols., quienes recolectaron una muestra de 300 pacientes con edades menores de 40 años con EVC isquémica en sus diversas variedades, de los cuales 32% fueron clasificados como criptogénicos, es decir, sin lograr identificar la etiología específica;15 es probable que algunos de estos casos pudieran corresponder a EF, pero dicha enfermedad no fue descartada.
En Argentina, un estudio multicéntrico llevado a cabo en 2017, recolectó los datos de 311 pacientes con EVC (80% infartos, 9% ataques isquémicos transitorios (AITs) y 11% hemorragias intracerebrales), de ellos sólo 1 caso presentó EF evidente con una mutación patógena: c.888G> A/p.Met296Ile/Exon 6, representando el 0.3% de la muestra y el 1% de los pacientes con infartos cerebrales criptogénicos.16
En 2017, el grupo de iniciativa Canadiense para el tamizaje de EF (Canadian Fabry Stroke Screening Initiative Study Group), de una cohorte de 365 pacientes con infarto cerebral y 32 con AIT, entre 18 y 55 años, identificaron un único caso portador de una variante genética de importancia desconocida (p.R118C) y sin variantes patogénicas bien reconocidas. Como resultado, si dicha variante se considera patógena, la prevalencia de EF sería del 0.3%.17 En suma, esto sugiere que deberían aplicarse métodos más rentables para el diagnóstico de EF, en lugar del cribado genético sistemático en esta población.
Fisiopatología
La EF se considera un trastorno ligado a X (Xq22.1), mutando al gen GLA que codifica para la proteína de la α-galactosidasa A (GLA, 300644)8 con un alto grado de penetrancia en hombres, e intermedia en las mujeres: alrededor del 50-70% de las mujeres con mutaciones en el gen tienen manifestaciones de EF, mientras que cerca del 100% de los hombres presentan complicaciones de la enfermedad10 El gen de la α-galactosidasa A tiene 12kb de largo, con siete exones y codifica una proteína precursora de 429 aminoácidos que se procesa a una glicoproteína de 370 aminoácidos que funciona como un homodímero. Se conocen 596 mutaciones descritas para este gen, de ellas, 416 son mutaciones de tipo sin sentido/de parada, 83 deleciones pequeñas, 19 deleciones grandes, 32 defectos de empalme, tres reordenamientos complejos y una inserción grande.7
El proceso de la enfermedad primaria comienza en la infancia, o incluso en la etapa de desarrollo fetal, sin embargo, a diferencia de muchas otras enfermedades por almacenamiento lisosómico, la mayoría de los pacientes permanecen clínicamente asintomáticos durante los primeros años de vida. En la EF, se cree que el almacenamiento lisosómico y la actividad deficiente de α-galactosidasa A en plasma y leucocitos, conduce a la acumulación de globotriaosilceramida y glucoesfingolípidos con disfunción celular, lo que desencadena una cascada de eventos que incluyen muerte celular, metabolismo energético comprometido, lesión de vasos pequeños, disfunción de canales de potasio activados en las células endoteliales, estrés oxidativo, maduración alterada de autofagosomas e isquemia tisular, dando como resultado una disfunción orgánica progresiva.18
Manifestaciones clínicas (Tabla 1)
1.- Manifestaciones clínicas iniciales
Los síntomas iniciales más frecuentes de EF son crisis episódicas de dolor, que duran de minutos a horas, y que afectan principalmente a los pies o las manos, en general precipitadas por el ejercicio, la fiebre o el calor, y modificadas por acetaminofén.19 Los mecanismos responsables de la producción de las crisis de dolor no se conocen bien, pero es posible que el almacenamiento de glucofosfolípidos dentro de las células endoteliales de los vasa nervorum, las células perineurales o la raíz dorsal y los ganglios autónomos, pueda causar una reactividad vasomotora alterada, lo que resulta en un estado hipóxico.11
Tabla 1 Características clínicas en Enfermedad de Fabry. Modificado de Germain D.
| Sistema | Signos y síntomas |
|---|---|
| Órganos sensitivos | -Oculares Cornea verticillata Catarata posterior Vasculopatía (retina, conjuntiva) -Auditivos (Vértigo/tinnitus) Sordera neurosensorial |
| Nervioso Central | -Deterioro cognitivo -Cefalea -EVC hemorrágica o isquémica con predominio circulación posterior-Trastornos psiquiátricos |
| Nervioso periférico | -Neuropatía dolorosa (predominantemente fibra pequeña)
Dolor neuropático (Acroparestesias) -Disautonomía Intolerancia al calor o frío Hipohidrosis o anhidrosis |
| Cardiovascular |
-Arritmias -Hipertrofia ventricular no explicada -Anormalidades de la conducción en elelectrocardiograma -Valvulopatías |
| Respiratorio | -Asma
-Disnea por reducción en capacidad de ejercicio |
| Digestivo | -Dolor abdominal postprandial -Náusea/vómito -Diarrea episódica-Saciedad temprana |
| Nefrourinario |
-Microalbuminuria/proteinuria -Hematuria -Síndrome nefrótico -Enfermedad renal de etiología no determinada |
| Piel | -Angioqueratomas
-Dishidrosis (hipo/anhidrosis) |
2.- Manifestaciones clínicas clásicas
Los pacientes con la forma clásica de la enfermedad (sin actividad residual de α-galactosidasa A), tienen anomalías dismórficas típicas, particularmente en la cara. Estos dismorfismos incluyen plenitud periorbitaria, crestas supraorbitarias prominentes, cejas pobladas, frente hundida, lóbulos prominentes de las orejas o rotación de las mismas, ángulo nasal pronunciado, nariz generosa/punta nasal bulbosa, puente nasal prominente, base alar ancha, labios carnosos, rasgos toscos y prognatismo.7 Durante la adolescencia se suman lesiones cutáneas llamadas angioqueratomas, que se localizan habitualmente a nivel periumbilical, genital y raíces de muslos.20 El acúmulo de material citoplasmático de aspecto lipoide provoca una deformación epitelial de los mechones glomerulares, de los túbulos, células endocapilares glomerulares, células musculares arteriolares que clínicamente se manifiesta como una proteinuria e insuficiencia renal.8 Dicha insuficiencia renal es la causa de muerte primaria en los pacientes con EF. 21
3.- Manifestaciones de la EF en sistema nervioso.
Las complicaciones neurológicas de la EF son frecuentes y afectan tanto al sistema nervioso central (SNC) como al sistema nervioso periférico(SNP).10 Los datos del registro mundial de EF, una gran cohorte de 2,446 pacientes, indican que los eventos de EVC son frecuentes en homocigotos y heterocigotos, ocurriendo en 6.9% y 4.3%, respectivamente;22 de estos, el 87% fueron isquémicos y el 13% fueron hemorrágicos.23 En el registro de Fabry, la mayoría de los pacientes que experimentaron una primera EVC lo hicieron entre los 20 y los 50 años, y 22% tuvieron una primera EVC antes de los 30 años.24 Las manifestaciones de la EF en del SNC incluyen: enfermedad vascular cerebral, deterioro auditivo con tinnitus, vértigo, trastornos psiquiátricos y deterioro cognitivo.10
3.1.- Manifestaciones clínicas de la EF como enfermedad vascular cerebral
Los hombres homocigotos pueden presentar disartria, diplopía, vértigo, nistagmo, náuseas y/o vómitos, hemiparesia, ataxia o síntomas sensitivos hemicorporales, en relación con la localización y tipo de EVC que desarrollen. La cefalea es bastante infrecuente, reportada sólo en el 20% de los pacientes. En la mayoría de los pacientes (58%), la presentación es compatible con isquemia del territorio vertebrobasilar, mientras que la circulación anterior fue definitivamente sintomática en aproximadamente el 20% de los pacientes.25 La demencia vascular por enfermedad penetrante de vasos pequeños también se ha descrito en pacientes con EF y debe tenerse en cuenta en la evaluación de una demencia inexplicable, en particular en hombres menores de 65 años.26,27
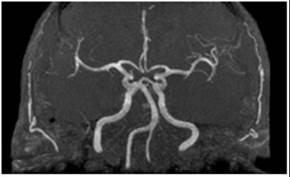
Las mujeres heterocigotas también pueden desarrollar síntomas de deterioro neurológico, siendo los más reportados el deterioro cognitivo, vértigo, ataxia, hemiparesia, síntomas sensitivos hemicorporales y cefalea. En la mitad de las pacientes, la presentación clínica fue compatible con afectación dentro del territorio vertebrobasilar, mientras que el territorio carotideo estaba definitivamente comprometido en sólo el 10% de los casos.25 También se han informado oclusiones de la arteria central de la retina 28 y oclusiones de la vena central de la retina.29 Además de la dolicoectasia, en los estudios de imagen se pueden observar lesiones de sustancia blanca y el “signo del pulvinar” caracterizado por hiperintensidad en la región posterior de tálamo, en la secuencia T1 de la resonancia magnética.9
Las características comunes del SNP incluyen: neuropatía periférica (especialmente neuropatía de fibras delgadas) con acroparestesias, disfunción autonómica caracterizada por hipohidrosis, dismotilidad intestinal y desregulación térmica y vasomotora periférica.10
3.2.- Hallazgos patológicos en sistema nervioso
Los hallazgos de la autopsia neuropatológica son consistentes con eventos previos de isquemia cerebral y, rara vez, hemorragia intracerebral; también se pueden observar infartos hemisféricos extensos y superficiales, múltiples infartos pequeños y profundos e infartos del tronco encefálico y/o cerebelo, este último grupo más frecuente en homocigotos y heterocigotos sintomáticos.25
Los vasos del polígono de Willis a menudo parecen engrosados. El estrechamiento de la luz y los depósitos intracelulares en arterias y arteriolas son hallazgos adicionales.19 La dolicoectasia de las arterias basilar y vertebral, y menos frecuente de las arterias carótidas, es un hallazgo constante tanto en homocigotos como en heterocigotos sintomáticos.25
Los eventos de EVC isquémica y hemorrágica en EF parece presentarse en una proporción similar a la observada en la población general, pero en edades más tempranas, y los AITs parecen ser un factor de riesgo de EVC. La hipertensión se ha considerado el factor de riesgo más importante de EVC en la EF, y su efecto probablemente se ve potenciado por la degeneración vascular subyacente secundaria al depósito de glucoesfingolípidos. En el Registro de Fabry, los pacientes con EVC tenían más probabilidades de informar antecedentes de hipertensión en comparación con los pacientes con EF sin EVC, 52.9% frente a 20.5%, respectivamente.9
Se han identificado diversas anomalías en el flujo sanguíneo cerebral y en las paredes de los vasos intracraneales, que pueden no ser exclusivas del sistema arterial. Varios mecanismos pueden contribuir a la vasculopatía de la EF, que incluyen: disfunción endotelial, desregulación de vías de óxido nítrico, estado protrombótico, hiperhomocisteinemia, niveles elevados de lípidos y moléculas de adhesión de leucocitos (Tabla 2).19
Tabla 2 Mecanismos del infarto cerebral en Enfermedad de Fabry. Modificado de Caplan, et al.
La dolicoectasia encontrada con frecuencia en la EF, particularmente en los grandes vasos de la circulación posterior, puede estar relacionada con el debilitamiento mecánico de la pared del vaso, causado por el depósito de glucoesfingolípidos y la hipertensión. Los mecanismos fisiopatológicos de la EVC asociados a la dolicoectasia incluyen la formación de émbolos y oclusión de arterias penetrantes del tallo cerebral. La afectación cardíaca en la EF también puede predisponer a la EVC, principalmente su asociación con arritmias.9
Diagnóstico (Figura 1)
En los hombres, el diagnóstico de EF se realiza midiendo la actividad de la enzima α-galactosidasa A en plasma o leucocitos periféricos. En contraste, las mujeres pueden tener los niveles de actividad de α-galactosidasa A en valores normales, por lo tanto, su medición no es de utilidad en aquellas mujeres con sospecha de EF por heterogocidad, y deben someterse a la determinación del genotipo de GLA. Los niveles de sustratos de α-galactosidasa A elevados en plasma y orina (Gb3 y liso-Gb3) sugieren el diagnóstico de EF. 30
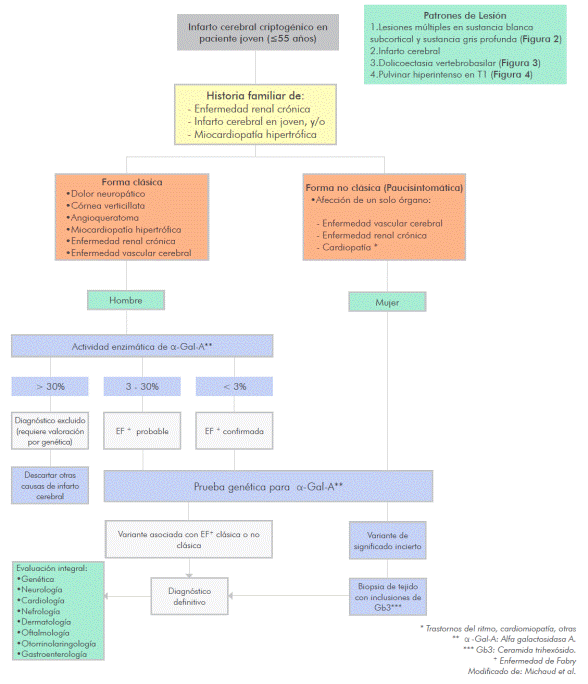
Las muestras requeridas para su diagnóstico en sangre son de 3ml de sangre completa en un tubo con EDTA (ácido etilenodiaminatetraacético). Actualmente se cuenta con el diagnóstico enzimático en gotas de sangre cuya muestra puede recolectarse y transportarse en papel de filtro, esta nueva metodología posibilita el envío de muestras a distancia para su análisis enzimático, diagnóstico retrospectivo y tamizaje poblacional.31
La secuenciación del gen GLA constituye el estándar de oro para el diagnóstico de EF. Debido a la herencia ligada a X, no hay aporte del gen mutado de padre a su descendencia, mientras que las mujeres heterocigotas tienen un riesgo del 50% de transmitir el gen mutado a su descendencia.30
Tamizaje de enfermedad de Fabry en enfermedad vascular cerebral
La detección de EF en poblaciones de alto riesgo se convirtió en una preocupación importante cuando estuvo disponible, desde 2001, el tratamiento de reemplazo enzimático (ERT), aplicado cada 2 semanas. Al respecto, se llevaron a cabo estudios en diferentes contextos que evidenciaban las complicaciones graves de la EF, incluida la enfermedad renal crónica, la hipertrofia ventricular izquierda (HVI) y la EVC. Cabe mencionar que el cribado puede estar sesgado hacia los pacientes con la enfermedad más grave y los fenotipos clásicos. En el Registro de Fabry, 22 los pacientes con EVC fueron diagnosticados más tarde que aquellos sin EVC, y la mayoría de ellos no habían experimentado eventos renales o cardíacos antes de su primer evento cerebral, lo que sugiere que las características clásicas de la enfermedad pueden estar ausentes o ser más sutiles en estos pacientes.
En tiempos recientes se han notificado fenotipos atípicos con una frecuencia creciente -algunos de ellos con EVC como característica de presentación-, en los que, debido a que su reconocimiento clínico requiere un alto índice de sospecha, el diagnóstico de EF a menudo se retrasa o se pasa por alto. Por lo tanto, se desconoce la verdadera prevalencia de EF en pacientes jóvenes con EVC.9
Respecto a la población latinoamericana, sólo se tiene información procedente de 333 pacientes incluidos en el registro mundial de EF,32 principalmente de Argentina, Chile, Colombia, Perú y, entre ellos, algunos mexicanos. Forman parte del registro 167 mujeres y 166 hombres latinoamericanos, con una edad promedio de 35.5 años para los hombres y 37.9 años para las mujeres. De estos pacientes, 8 hombres (5%) y 3 mujeres (2%) presentaron una EVC. La mayor parte de los pacientes latinoamericanos de este registro provienen de servicios de nefrología y cardiología. Al momento, no hay actualización de datos por parte del registro internacional de EF, cuyo reclutamiento y registro se encuentra abierto.22
El diagnóstico y tamizaje de la EF ha sido objeto de estudio principalmente en países de altos ingresos económicos, incluyendo programas de tamizaje neonatal. En países como Dinamarca, Australia y Japón, se han implementado programas de detección temprana que se basan en obtención de muestra de sangre seca en papel filtro (DBS, del inglés Dried Blood Sample), generalmente obtenida entre las 24 y 72 horas de vida extrauterina, y que consiste en medir la actividad de la enzima α-galactosidasa A o acumulación de globotriaosilceramida.33 Esos trabajos han logrado determinar una sensibilidad del 100% para la detección de recién nacidos con EF, pero con una especificidad variable cuando se compara con el patrón de referencia que es la secuenciación genética (con un valor predictivo positivo del 33 al 42%).34 En el caso de las mujeres la prueba de actividad enzimática en sangre tiene baja sensibilidad.
Tratamiento
En 2001, se aprobaron dos enzimas recombinantes para su uso en la enfermedad de Fabry: agalsidasa alfa (Replagal®, Shire/Takeda) y agalsidasa beta (Fabrazyme®, Sanofi Genzyme), denominadas terapia de reemplazo enzimático (ERT),35 las cuales demostraron reducción en la producción de HVI y reducción del avance de la enfermedad renal cuando la depuración se encontraba menor a 60 ml/min/1.73 m2, posiblemente atribuida a su terapia anti-proteinurica, lo cual podría reducir a largo plazo la incidencia de EVC al disminuir los factores de riesgo que la promueven tanto en hombres como mujeres. En la misma línea, Migalastat (1-desoxigalactonojirimicina; Galafold, Amicus Therapeutics) fue aprobado en Europa en mayo de 2016, en Canadá en septiembre de 2017, en Japón en marzo de 2018, y en los EEUU en agosto del mismo año, para el tratamiento a largo plazo de la EF en adultos (≥18 años de edad en Estados Unidos y Canadá, ≥16 años en otros países) con una mutación susceptible y una tasa de filtración glomerular estimada (eGFR) ≥30 mL/min/1.73 m2. El Migalastat administrado por vía oral es un pequeño acompañante que estabiliza la enzima α-Galactosidasa A endógena y apoya un plegamiento de proteínas adecuado en el retículo endoplásmico, lo que conduce a una mayor actividad enzimática de la misma y estabilidad en los lisosomas de pacientes portadores de una mutación susceptible.36 En un estudio multicéntrico del uso de Migalastat por 12 meses (estudio FAMOUS), se demostró reducción de la masa del ventrículo izquierdo, aunque no se ha logrado reducir el avance de la enfermedad renal, posiblemente por la intervención de otros factores detonantes.37
En el manejo de un infarto cerebral agudo en pacientes con EF, se puede considerar trombolisis intravenosa 38 o abordaje endovascular; la experiencia con el uso de cualquiera de los dos en este entorno es de limitada a inexistente, aunque no hay razones específicas para negar dichos tratamientos.10
En la prevención secundaria, el tratamiento está lejos de ser satisfactorio, ya que no se dispone de una terapia específica para las complicaciones cerebrovasculares de la EF. La administración de agentes antiplaquetarios puede ayudar a prevenir los efectos ateroscleróticos y tromboembólicos del daño al endotelio vascular, pero la experiencia con este enfoque es escasa.10 De igual modo, los agentes anticoagulantes deben considerarse para ayudar a prevenir recurrencias de infarto cerebral cuando la causa implicada es embolismo cardiaco.19
Pronóstico
La EF es una patología progresiva con una expectativa de vida reducida; la edad de sobrevida media para los hombres es de 50-55 años, y de 70 años para las mujeres. La calidad de vida se ve afectada en todos los pacientes, no sólo debido a daño a órgano blanco, sino también como resultado de otros síntomas que incluyen problemas gastrointestinales, acroparestesias, depresión e intolerancia a ciertas temperaturas. Desde su introducción en 2001, la terapia de reemplazo enzimático ha demostrado ser efectiva para el alivio de muchos de estos síntomas, así como para retrasar e incluso revertir la progresión de la enfermedad.7,39
Conclusión
Se recomienda que a todos aquellos pacientes jóvenes (<55años) con antecedente de uno o más eventos de EVC isquémica o hemorrágica, de etiología indeterminada (criptogénica), principalmente aquellos con enfermedades sistémicas (dermatológicas, cardiacas o renales), se les realice tamizaje de Enfermedad de Fabry, mediante la determinación de α-galactosidasa A o secuenciación del gen GLA. El tratamiento de la EVC en estos pacientes es similar a lo establecido en guías clínicas para población general, ya que no existe tratamiento específico para esta patología, pero requiere abordaje 40 y seguimiento multidisciplinario (véase Figura 2 en la siguiente página).
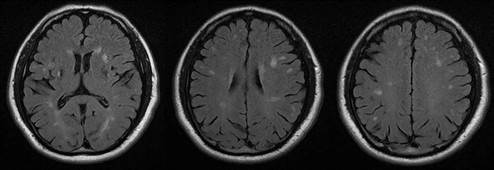
Figura 2 RM T2-FLAIR Axial. Lesiones hiperintensas multiples en sustancias blanca subcortical y sustancia gris profunda bilateral