Articles
Non-covalent Interactions in Dihalogenated Compounds Ch(C6H4CH2X)2 (Ch = O, S; X = Cl, Br, I). Synthesis, Crystal Structure, and Hirshfeld Surface Analysis

-
Publication dates-
March 24, 2025
Apr-Jun , 2024
- Article in PDF
- Article in XML
- Automatic translation
- Send this article by e-mail
- Share this article +
Abstract.
In this work, the synthesis and structural study by means of single-crystal X-ray diffraction of compounds of general formula Ch(C6H4CH2 X)2 (Ch = O, S; X = Cl, Br, I) is reported. These compounds contain two flexible hydrocarbonated arms -CH2-X in the ortho positions to the Ch heteroatom. These compounds were synthesized through a linear synthesis starting from diphenylether or diphenylsulfide. Based on the structural analysis, we describe the more relevant molecular features as well as the non-covalent interactions that the heavy halogen atoms display with other moieties that promote the cohesion of the crystal arrangement. The Hirshfeld analysis displayed that the X···π, X···X, and C-H···X interactions are quite significant in the crystal arrangement.
Keywords::
Non-covalent interactions, dihalogenated compounds, Hirshfeld surfaces
Introduction
In many aspects, the physicochemical properties and reactivity of halogenated organic compounds is strongly based on the nature of the X halogen substituent (X = F, Cl, Br, or I); for example, with an increase in the atomic number of the halogen, C-X bond energies decrease markedly, i.e., C-F > C-Cl > C-Br > C-I [1], and the order of reactivity can generally be predicted by the relative easiness with which the respective carbon-halogen bonds are broken. Other features such as the electron-withdrawing effect, atomic size, and polarizability of the halogen substituent also affect the reactivity of these halogenated compounds [2]. A typical reaction of these compounds is nucleophilic substitution of the halogen by electron-rich molecules or anions such as ammonia, cyanide (a pseudohalide), hydroxide, alkoxides, or even another halogen [3]. In this vein, halogen-by-halogen exchange substitution provides a convenient route for synthesizing alkyl halides [4]. Other typical and very useful reaction of alkyl halides involves the formation of free radicals in the presence of very active alkaline or alkaline earth metals; subsequent reaction of these radicals can lead to the formation of organometallic compounds or coupling of the alkyl radicals to enlarge carbon chains [5].
-
1Lange's Handbook of Chemistry, 1999
-
2Halogenated Organic Compounds - A Global Perspective. In Dehalogenation, 2004
-
3Halides, Pseudo‐Halides and Azides; Part 1 and Part 2 in The Chemistry of Functional Groups, 1983
-
4Halogen Chemistry. In Encyclopedia of Physical Science and Technology, 2003
-
5Green sustainable process for chemical and environmental engineering and science: A. Sonochemical protocols for Grignard reactions, 2020
On the other hand, the presence of a halogen atom makes possible the formation of either Y-X···A intra- or intermolecular non-covalent interactions; here, Y-X is the halogen bond donor such as I2, Br2, ICl, haloalkanes; haloarenes, haloimides, etc., meanwhile A represents the halogen bond acceptor that can be an atom with an electronic lone pair, a p system, or an anion [6,7]. Moreover, the halogen atom usually displays two regions; an electrophilic region diametrically opposed to the Y-X sigma bond (named as a σ-hole) and a nucleophilic region transversal to the stated bond (Fig. 1). Hence, due to the anisotropic distribution of the electron density, these two interactions have an important electrostatic component. [8]
-
6NobelPrize.org, 2023
-
7Pure Appl. Chem, 2013
-
8Chem. Rev, 2016
Thumbnail
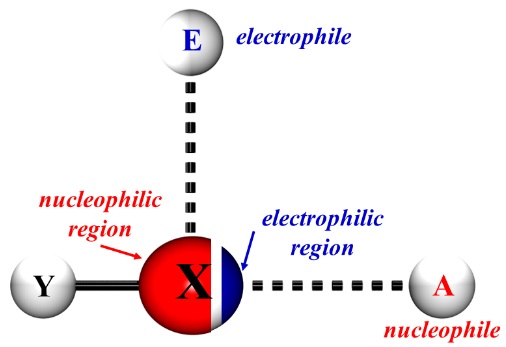
Fig. 1
Diagrammatic representation of an X···A halogen bond; also, is displayed a possible transversal E···X interaction.
Diagrammatic representation of an X···A halogen bond; also, is displayed a possible transversal E···X interaction.
A chief key to the formation halogen bonds is the polarizability of the halogen atom. Therefore, the strongest halogen bonds are formed by the most easily polarizable halogens, and the strength of the halogen bonds typically decreases in the order I > Br > Cl > F [9]. Several distinctive features of X···A halogen bond, such as the high directionality and strength tunability, prompted a plethora of applications in organocatalysis [10], crystal engineering [11]and drug design [12], and their influence on packing modes and the consequent orientation of organic molecules, are also considered important in conjunction with the practically ubiquitous both classical and no-classical hydrogen bond [13,14].
-
9J. Am. Chem. Soc, 2005
-
10J. Am. Chem. Soc, 2019
-
11Halogen Bonding in Crystal Engineering: Recent Advances in Crystallography, 2012
-
12Chem. Phys. Lett, 2021
-
13Cryst. Eng, 2000
-
14J. Mol. Struct, 2022
Thus, as a part of our studies in the reactivity of halogenated compounds [15], herein we report the structural analysis in crystalline state of two series of conformationally flexible di(2-halomethylphenyl)ethers (compounds 3a, 4a, and 5a) and di(2-halomethylphenyl)thioethers (compounds 3b, 4b, and 5b) of general formula Ch(C6H4CH2 X)2, where Ch is a chalcogen atom (oxygen or sulfur) and X = Cl, Br or I. To provide a structural context, in the 3D pie chart showed in Fig. 2 are presented the results of a delimited search in the Cambridge structural database for compounds containing at least one C6-CH2-X moiety.
-
15Inorg. Chem. Commun, 2018
Thumbnail
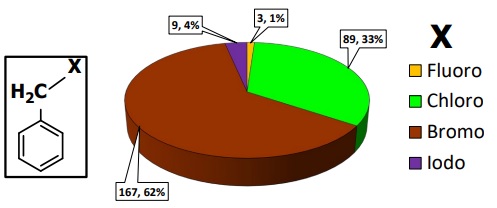
Fig. 2
Numerical and relative proportion of halocompounds containing at least one C6-CH2-X moiety. (268 hits; data retrieved from CSD version 5.43, updated to November 2022).
Numerical and relative proportion of halocompounds containing at least one C6-CH2-X moiety. (268 hits; data retrieved from CSD version 5.43, updated to November 2022).
From these data, it is clear the dominance of the bromomethylenic compounds (167 compounds), and the scarceness of fluoro- and iodo- ones. In addition, we want to highlight two experimental facts: the first one is that the presence of two conformationally flexible -CH2-Br arms in 1,4-dibromo-2,5-bis(bromomethyl)benzene allowed dimorphism, displaying Br···Br non-covalent interactions in solid crystalline state [16]. The second one is that there is just one compound with a slightly similar chemical environment to 3a-5a and 3b-5b, reported as a private communication (Ref. CSD code MUMQAO) [17].
-
16Cryst. Eng Comm, 2016
-
17CSD Communication (Private Communication), 2015
In the title compounds, the presence of two -CH2-X arms of different nature in both series prompted the formation of several X···p and X···X alongside to C-H···X interactions. A Hirshfeld surface analysis was carried out to describe the interactions present in the isostructural compounds.
Experimental
Instrumentation and software
Melting points were determined with a Mel-Temp II instrument. IR spectra were recorded in the 4,000-400 cm−1 range on a Perkin-Elmer System 2000 FT-IR spectrometer, as KBr pellets. 1H and 13C NMR spectra were recorded in CDCl3 on a Bruker Avance III 400 MHz spectrometer (400.1 MHz for 1H and 100.0 MHz for 13C) at 23 °C and calibrated by using the residual proton resonance of CDCl3. Chemical shifts (δ) are reported in ppm and coupling constants (J) are in Hz. For the assignment we used 2D correlation experiments such as COSY, HSQC and HMBC.
Suitable crystals for the X-ray diffraction analysis were obtained for compounds 3a, 4a, 5a, 4b, and 5b by slow evaporation of CH2Cl2 solutions. Each single crystal was either glued on a tip of a thin glass fiber or mounted on a Nylon loop. Diffraction data were collected at 294 K in an Agilent Gemini diffractometer equipped with an Atlas CCD detector; we used graphite-monochromated Mo-Kα radiation (0.71073 Å). Absorption corrections were made by multi-scan methods using the CrysAlisPro software [18]. By using the Olex2 interface [19], the structures were solved by intrinsic phase methods using SHELXT [21] and all the non-hydrogen atoms were anisotropically refined by full-matrix least-squares on F2 using SHELXL [22]. Hydrogen atoms were placed in their calculated positions and then refined using the riding model. Crystallographic data are listed in Table 1, and selected bond distances and angles are given in Table 2. The asymmetric unit of 3a displayed just the half of the molecule (see gray moiety in Fig. 3); the other half is related by a two-fold axis, where the oxygen atom is placed on it (see green moiety in Fig. 3). The ORTEP views of the molecular structures are shown in Figures 3 and 4.
-
18Oxford Diffraction CrysAlis software system, 2014
-
19Appl. Cryst, 2009
-
21Acta Cryst. C, 2015
-
22CrystEngComm, 2009
Thumbnail
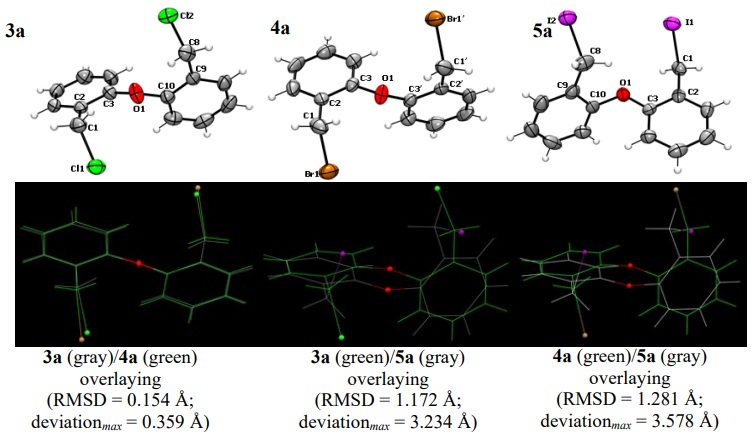
Fig. 3
Molecular structures of O(C6H4CH2 X)2 compounds (ORTEP at 50 %) and overlay results of the corresponding pairs.
Molecular structures of O(C6H4CH2 X)2 compounds (ORTEP at 50 %) and overlay results of the corresponding pairs.
Thumbnail
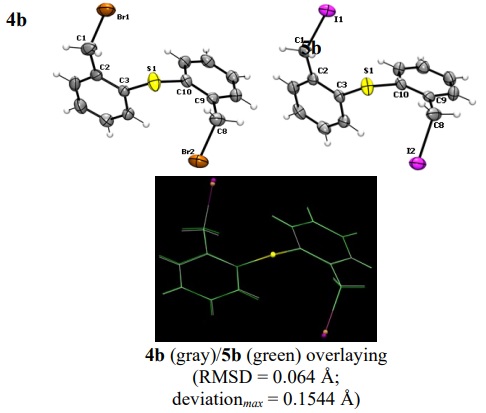
Fig. 4
Molecular structures of S(C6H4CH2 X)2 compounds (ORTEP at 50 %) and overlay results of the pair.
Molecular structures of S(C6H4CH2 X)2 compounds (ORTEP at 50 %) and overlay results of the pair.
Table 1
Crystal data and refinement details.
Crystal data and refinement details.
| 3a | 4a | 5a | 4b | 5b | |
| Empirical formula | C14H12C12O | C14H12Br2O | C14H12I2O | C14H12Br2S | C14H12I2S |
| Formula weight [g/mol] | 267.14 | 356.06 | 450.04 | 372.12 | 466.10 |
| Crystal system | triclinic | monoclinic | monoclinic | triclinic | triclinic |
| Space group | P−1 | I2/a | P21 | P−1 | P−1 |
| a [Å] | 7.6808(4) | 12.279(3) | 8.2871(4) | 8.2610(14) | 8.2734(5) |
| b [Å] | 7.9888(4) | 9.2954(17) | 4.6487(3) | 9.2086(13) | 9.5171(7) |
| c [Å] | 10.7391(4) | 11.972(3) | 18.5371(12) | 10.6912(17) | 10.7511(9) |
| α [º] | 87.075(4) | 90 | 90 | 102.043(13) | 103.115(7) |
| β [º] | 79.474(4) | 99.17(2) | 90.603(5) | 101.772(14) | 101.149(6) |
| γ [º] | 83.979(4) | 90 | 90 | 113.586(15) | 112.203(6) |
| V [Å3] | 643.95(5) | 1349.1(5) | 714.09(7) | 690.4(2) | 725.83(10) |
| Z | 2 | 4 | 2 | 2 | 2 |
| rcalc [g/cm3] | 1.378 | 1.753 | 2.093 | 1.790 | 2.133 |
| Absorption coefficient [mm-1] | 0.484 | 5.990 | 4.386 | 5.996 | 4.453 |
| F(000) | 276 | 696.0 | 420.0 | 364.0 | 436.0 |
| Crystal size [mm3] | 0.3 × 0.2 × 0.1 | 0.3 × 0.3 × 0.2 | 0.2 × 0.2 × 0.1 | 0.5 × 0.5 × 0.5 | 0.2 × 0.1 × 0.1 |
| 2Q range data collection [º] | 6.068 to 59.062 | 6.722 to 59.244 | 6.562 to 59.278 | 6.858 to 59.262 | 6.85 to 59.148 |
| Measured reflections | 23203 | 5234 | 18168 | 5913 | 5595 |
| Data/restraints/parameters | 3407/0/154 | 1678/0/78 | 3656/1/154 | 3262/0/154 | 3432/0/155 |
| R1; wR2 [I>2(I)] | 0.0416, 0.0900 | 0.0442; 0.1146 | 0.0279; 0.0528 | 0.0458; 0.0795 | 0.0609; 0.0871 |
| R1; wR2 [All data] | 0.0680, 0.1026 | 0.0975; 0.1381 | 0.0417; 0.0578 | 0.1054; 0.1016 | 0.1339; 0.1079 |
| Goodness-of-fit on F2 | 1.031 | 1.040 | 1.039 | 0.995 | 1.031 |
| Largest diff. peak / hole [eÅ-3] | 0.27/−0.30 | 0.63/−0.57 | 0.46/−0.52 | 0.33/−61 | 1.77/−1.32 |
| Flack parameter | Not applicable | Not applicable | −0.019(16) | Not applicable | Not applicable |
| CCDC | 2252394 | 2252390 | 2252392 | 2252393 | 2252391 |
Table 2
Selected bond lengths (Å) and angles (°) for Ch(C6H4CH2 X)2.
Selected bond lengths (Å) and angles (°) for Ch(C6H4CH2 X)2.
| 3a | 4a | 5a | 4b | 5b | |
| (Ch = O X = Cl) | (Ch = O X = Br) | (Ch = O X = I) | (Ch = S X = Br) | (Ch = S X = I) | |
| C–X | 1.801(2); 1.802(2) | 1.950(4) | 2.180(6); 2.150(6) | 1.971(4); 1.968(4) | 2.170(5); 2.163(5) |
| C–Ch | 1.383(2); 1.385(2) | 1.381(4) | 1.368(6); 1.408(6) | 1.783(4); 1.794(3) | 1.776(4); 1.769(4) |
| C–Ch–C | 119.22(14) | 118.5(4) | 118.9(4) | 101.03(17) | 101.6(2) |
| C2–C3–Ch–C10 | 140.64(16) | 138.6(4) | 179.0(5) | 134.9(3) | 137.0(4) |
| C9–C10–Ch–C3 | 144.21(16) | 138.6(4) | 102.3(6) | 127.3(3) | 126.8(4) |
| |Δta| | 3.57 | 0.0 | 76.7 | 7.6 | 10.2 |
The atomic coordinates of all 3a-5a diphenylether and 4b and 5b diphenylthioether compounds determined by X-ray crystallographic studies at 294 K were used to construct the molecular Hirshfeld surfaces based on the electron distribution calculated as the sum of spherical atomic electron densities (the promolecule), which dominates the corresponding sum over the crystal (the procrystal) yielding an implicit 0.5 value for the promolecule-to-procrystal ratio. The bond lengths involving hydrogen atoms were adjusted to values derived from neutron diffraction [22]. The Hirshfeld surfaces mapped with d norm and 2D fingerprint plots presented were generated using CrystalExplorer21 with a high standard surface resolution [23]. They were mapped using a fixed color scale, red-white-blue, where red highlights are for contacts shorter than van der Waals (vdW) radii sum, white for contacts around vdW separation, and blue is for contacts longer than the vdW sum.
-
22CrystEngComm, 2009
-
23J Appl. Cryst, 2021
The molecular electrostatic potential mapping (MEP) was calculated to characterize the close contacts by projecting the Hirshfeld surface with this MEP. We used the Tonto quantum modelling package [24] implemented in CrystalExplorer21 using the Becke three-parameter Lee-Yang-Parr (B3LYP) hybrid functionals [25] with the DGDZVP basis set [26,27].
-
24Comput. Sci. ICCS, 2003
-
25J. Chem. Phys, 1993
-
26J. Chem, 1992
-
27J. Phys. Chem, 1992
General methods
All manipulations involving n-BuLi reagent were performed in a dry nitrogen atmosphere using standard Schlenk techniques. Solvents, N,N,N´N´-tetramethylethylenediamine (TMEDA), and N,N-dimethylformamide (DMF) were purchased from Sigma-Aldrich and dried by standard methods and distilled prior to use. The reagents used in the syntheses were purchased from Aldrich and used without purification. Merck Kiesel gel 60 (0.063-0.40 mm) was used for column chromatography.
The sequence of the reaction steps is summarized in Scheme 1. Dihalogenated compounds 3a, 3b, 4a, 4b, 5a and 5b were prepared by linear synthesis, starting from either diphenylether or diphenylthioether, respectively (the a letter refers to the compounds with Ch = oxygen and b to compounds with Ch = sulfur).
Thumbnail
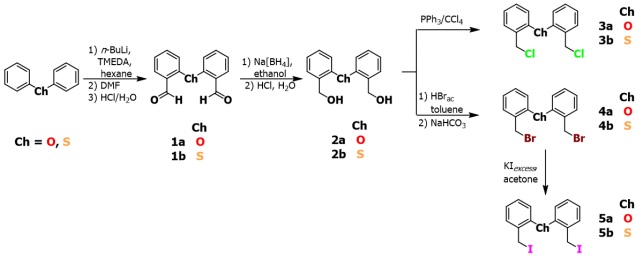
Scheme 1
Synthesis of dihalogenated compounds.
Synthesis of dihalogenated compounds.
Synthesis of the dialdehyde O(C6H4CHO)2 (1a)
Dialdehyde 1a is known from the literature [28, 29]. Modification of this method allowed the desired product to be obtained in a much higher yield and without purification by column chromatography. Diphenylether (6.5 mL, 41 mmol) was dissolved in dry hexane (40 mL) under an inert nitrogen atmosphere, and TMEDA (13.5 mL, 90.2 mmol) was added at 25 °C. A solution of n-butyllithium in hexane (2.5 M, 36.1 mL, 90.2 mmol) was added dropwise to the stirred reaction mixture at 0 °C. This mixture was stirred at 0 °C for 22 h, turning into a beige one. Then, dry DMF (11.5 mL, 147.6 mmol) was poured dropwise into the reaction mixture. The white solution was stirred for 1 h at 70 °C. Aqueous hydrochloric acid (40 mL HCl in 60 mL of distilled water) was added, and the mixture was stirred for fifteen minutes. After, the compound was extracted with CHCl3 (30 mL). The combined organic phases were dried with Na2SO4, and the volatiles were removed under reduced pressure to yield the title compound as a beige solid (8.8 g, 95 %).
-
28Bull. Chem. Soc. Jpn, 1991
-
29Inorg. Chem, 2003
mw = 226.22 g/mol. mp = 65 °C. IR (KBr, cm−1): ν = 3071, 2856, 2755, 1685, 1599, 1472, 1455, 1394, 1276, 1224, 1097, 881, 827, 758. 1H-RMN (400 MHz, CDCl3, ppm) δ = 10.50 (s, 2H, H1), 7.99 (d, 3 J = 7.69 Hz, 2H, H7), 7.59 (t, 3 J = 7.69 Hz, 2H, H5), 7.30 (t, 3 J = 7.80 Hz, 2H, H6), 6.96 (d, 3 J = 8.32 Hz, 2H, H4). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 188.7 (C1), 158.9 (C3), 136.1 (C5), 129.3 (C7), 127.3 (C2), 124.6 (C6), 119.2 (C4).
Synthesis of O(C6H4CH2OH)2 (2a)
The diol 2a is known from the literature [30]; here is introduced an alternative procedure to obtain it in a much higher yield. Thus, to an ethanolic solution (60 mL) of 1a (8.8 g, 38.9 mmol) was slowly added Na[BH4] (3.8 g, 97.4 mmol), and the resulting mixture was stirred for 1 h at 0 °C and three hours at room temperature. Then, it was acidified with diluted HCl (40 mL HCl in 60 mL of distilled water), followed by extraction with CH2Cl2 (60 mL). The combined organic phases were washed with brine (60 mL), dried (Na2SO4), and the volatiles were removed under reduced pressure. The product was obtained as a white solid (9.35 g, 99 %).
-
30Polyhedron, 2012
mw = 230.25 g/mol. mp = 86 °C. IR (KBr, cm−1): ν = 3272 (OH), 3082, 3039, 2929, 2886, 1689, 1604, 1581, 1484, 1452, 1230, 1110, 1034, 880, 755. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.39 (d, 3 J = 7.19 Hz, 2H, H7), 7.25 (td, 3 J = 7.69 Hz, 4 J = 1.22 Hz, 2H, H5), 7.10 (td, 3 J = 7.44 Hz, 4 J = 0.69 Hz, 2H, H6), 6.82 (d, 3 J = 8.06 Hz, 2H, H4), 4.68 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 154.8 (C3), 131.4 (C7), 130.1 (C5), 129.3 (C2), 123.7 (C6), 117.9 (C4), 61.2 (C1).
Synthesis of the dichlorinated compound [{O(C6H4CH2)2}Cl2] (3a)
To a reaction flask, were added the diol 2a (1.0 g, 4.3 mmol), carbon tetrachloride (20 mL), and triphenylphosphine (2.73 g, 10.4 mmol). The solution was heated at 65-70 °C for 24 h, and the reaction mixture was cooled and filtered. The yellow solution was chromatographed over a silica gel column with hexane/dichloromethane (1:1). The solution was evaporated to give a colorless crystalline solid (0.50 g, 43%).
mw = 267.14 g/mol. mp = 68 °C. IR (KBr, cm−1): ν = 3036, 2922, 2851, 1581, 1483, 1452, 1239, 1193, 1104, 914, 748, 672. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.50 (dd, 3 J = 7.60 Hz, 4 J = 1.50 Hz, 2H, H7), 7.28 (td, 3 J = 7.60 Hz, 4 J = 1.5 Hz, 2H, H5), 7.14 (t, 3 J = 7.40 Hz, 2H, H6), 6.83 (d, 3 J = 8.00 Hz, 2H, H4), 4.73 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 154.9 (C3), 131.2 (C7), 130.3 (C5), 128.7 (C2), 124.1 (C6), 118.5 (C4), 41.2 (C1).
Synthesis of dibrominated compound [{O(C6H4CH2)2}Br2] (4a)
Dibrominated compound 4a was synthesized according with the literature procedure [30]. To a toluene solution (30 mL) of 2a (9.35 g, 40.6 mmol), was added dropwise HBr (15.3 mL, 142.1 mmol) dissolved in toluene (10 mL), and the resulting mixture was refluxed for 22 h. Then, saturated aqueous NaHCO3 solution (20 mL) was carefully added, followed by extraction with CH3Cl3 (20 mL). The organic phase was separated, dried (Na2SO4), and evaporated to obtain a brown solid. The compound was purified by washing it with cold ethanol, to obtain a white crystalline solid (6.37 g, 44 %).
-
30Polyhedron, 2012
mw = 356.04 g/mol. mp = 92 °C. IR (KBr, cm−1): ν = 3058, 3034, 2985, 1579, 1485, 1449, 1242, 1185, 899, 775, 605. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.49 (d, 3 J = 7.60 Hz, 2H, H7), 7.28 (t, 3 J = 8.00 Hz, 2H, H5), 7.13 (t, 3 J = 7.60 Hz, 2H, H6), 6.85 (d, 3 J = 9.20 Hz, 2H, H4), 4.66 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 154.9 (C3), 131.5 (C7), 130.3 (C5), 129.0 (C2), 124.1 (C6), 118.6 (C4), 28.2 (C1).
Synthesis of diiodo compound [{O(C6H4CH2)2}I2] (5a)
Preparation of 5a was based on literature methods where the starting materials are also primary alcohols [31, 32]. Thus, KI (3.5 g, 21.0 mmol) was added to 4a (3 g, 8.4 mmol) previously dissolved in acetone (40 mL). This mixture was refluxed for 22 h. Next, the solvent was removed in vacuo, and the residue was dispersed in CH2Cl2 (50 mL). The resulting precipitate was filtered off, and the solvent was removed in vacuo to yield 5a as a brown solid (3.7 g, 97 %).
-
31Tetrahedron, 2005
-
32J. Acta Crystallogr. E: Crystallogr. Commun, 2015
mw = 450.0 g/mol. mp = 96 °C. IR (KBr, cm−1): ν = 3034, 2972, 1577, 1484, 1450, 1243, 1150, 894, 755. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.45 (dd, 3 J = 7.62 Hz, 4 J = 1.63 Hz, 2H, H7), 7.22 (td, 3 J = 7.69 Hz, 4 J = 1.68 Hz, 2H, H5), 7.07 (td, 3 J = 7.52 Hz, 4 J = 0.98 Hz, 2H, H6), 6.81 (d, 3 J = 8.18 Hz, 2H, H4), 4.60 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 154.27 (C3), 131.0 (C7), 130.4 (C5), 129.7 (C2), 124.1 (C6), 118.6 (C4), -0.03 (C1).
Synthesis of the dialdehyde S(C6H4CHO)2 (1b)
1b was prepared in a similar way as 1a [29]. For the formation of the dilithiated compound, we used diphenylsulfide (5.0 mL, 30 mmol), dry hexane (40 mL), TMEDA (10.4 mL, 69.0 mmol), and n-butyllithium in hexanes (2.5 M, 36.1 mL, 90.2 mmol). Then, this mixture was refluxed for 40 minutes forming an orange solution. The reaction was cooled down to 25 °C before dry DMF (8.1 mL, 105 mmol) was poured dropwise into the reaction mixture. The white solution was stirred for 1 h. Aqueous hydrochloric acid (30 mL HCl in 60 mL of distilled water) was added, and the mixture was stirred for fifteen minutes, followed by extraction with CH3Cl (30 mL). The combined organic phases were dried (Na2SO4), and the volatiles were removed under reduced pressure to yield the 1b as a brown viscous liquid (6.5 g, 90 %).
-
29Inorg. Chem, 2003
mw = 242.29 g/mol. IR (KBr, cm−1): ν = 3059, 2850, 2738, 1688, 1584, 1458, 1440, 1259, 1198, 1040, 845, 822, 747. 1H-RMN (400 MHz, CDCl3, ppm) δ = 10.34 (s, 2H, H1), 7.95 (d, 3 J = 7.42 Hz, 2H, H7), 7.47 (m, 4H, H5), 7.16 (d, 3 J = 7.68 Hz, 2H, H6). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 191.7 (C1), 138.7 (C3), 135.1 (C2), 134.8 (C4), 132.7 (C7), 131.9 (C5), 128.0 (C6).
Synthesis of the diol S(C6H4CH2OH)2 (2b)
2b was prepared in a similar was as the 2a compound. 1b (6.5 g, 27 mmol), ethanol (40 mL), Na[BH4] (2.45 g, 64.8 mmol), diluted HCl (10 mL HCl in 20 mL of distilled water), CH2Cl2 (40 mL), brine (60 mL). The product was a yellow viscous liquid (6.6 g, 99 %).
mw = 246.32 g/mol. IR (KBr, cm−1): ν = 3376, 3283, 3057, 2884, 1613, 1442, 1198, 1031, 747. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.45 (dd, 3 J = 7.50 Hz, 4 J = 0.82 Hz, 2H, H7), 7.27 (td, 3 J = 7.37 Hz, 4 J = 1.38 Hz, 2H, H6), 7.18 (td, 3 J = 7.30 Hz, 4 J = 1.54 Hz, 2H, H5), 7.12 (dd, 3 J = 7.73 Hz, 4 J = 1.28 Hz, 2H, H4), 4.72 (s, 4H, H1), 2.92 (s, 2H, OH). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 141.3 (C3), 133.3 (C2), 132.2 (C4), 128.8 (C7), 128.6 (C5), 127.9 (C6), 63.4 (C1).
Synthesis of the dichlorinated compound [{S(C6H4CH2)2}Cl2] (3b)
3b was prepared in a similar was as 3a. 2b (1.5 g, 6.2 mmol), carbon tetrachloride (20 mL), triphenylphosphine (3.6 g, 13.6 mmol). The yellow solution was chromatographed over a silica gel column with hexane/dichloromethane (1:1). The compound is a yellow liquid (0.77 g, 44 %).
mw = 267.14 g/mol. IR (KBr, cm−1): ν = 3057, 2956, 2850, 1589, 1468, 1440, 1263, 1205, 1041, 821, 736, 666. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.51 (dd, 3 J = 7.50 Hz, 4 J = 1.10 Hz, 2H, H7), 7.30 (td, 3 J = 7.80 Hz, 4 J = 1.30 Hz, 2H, H6), 7.24 (td, 3 J = 7.40 Hz, 4 J = 1.30 Hz, 2H, H5), 7.16 (dd, 3 J = 7.71 Hz, 4 J = 0.90 Hz, 2H, H4), 4.79 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 138.2 (C3), 135.0 (C2), 132.9 (C4), 130.7 (C7), 129.7 (C5), 128.2 (C6), 44.5 (C1)
Synthesis of dibrominated compound [{S(C6H4CH2)2}Br2] (4b)
Dibrominated compound 4b was synthesized according with the literature process [30]. To a toluene solution (20 mL) of diol 2b (6.6 g, 26.79 mmol) was added dropwise HBr (10.8 mL, 93.76 mmol) dissolved in toluene (10 mL) and the resulting mixture was stirred for 22 h at reflux temperature. Then saturated aqueous NaHCO3 solution (20 mL) was added carefully, followed by extraction with CH3Cl (20 mL). The organic phase was separated, dried (Na2SO4) and evaporated to obtain a brown liquid. For the purification of the compound 4b, a chromatographic column of the crude reaction is carried out, using silica as stationary phase and dichloromethane as eluent. From the purification a light-yellow viscous liquid was obtained, to crystallize the product were seeded crystals (2.5 g, 25 %).
-
30Polyhedron, 2012
mw = 372.11 g/mol. mp = 71 °C. IR (KBr, cm−1): ν = 3051, 2973, 1563, 1469, 1440, 1218, 1038, 764, 730, 606. 1H-RMN (400 MHz, CDCl3, ppm) δ = 7.47 (dd, 3 J = 7.54 Hz, 4 J = 1.35 Hz, 2H, H7), 7.27 (td, 3 J = 7.07 Hz, 4 J = 1.55 Hz, 2H, H6), 7.22 (td, 3 J = 7.26 Hz, 4 J = 1.46 Hz, 2H, H5), 7.16 (dd, 3 J = 7.71 Hz, 4 J = 1.35 Hz, 2H, H4), 4.71 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 138.5 (C3), 135.2 (C2), 133.1 (C4), 131.2 (C7), 129.7 (C5), 128.2 (C6), 31.8 (C1).
Synthesis of diiodo compound [{S(C6H4CH2)2}I2] (5b)
Preparation of the title compounds was based on literature methods [31,32]. KI (1.12 g, 6.72 mmol) was added to 4b (1.0 g, 2.69 mmol) dissolved in acetone (25 mL). This mixture was heated to reflux for 22 h. After the solvent was removed in vacuo, the residue was dispersed in CH2Cl2 (30 mL). The resulting precipitate was filtered off, and the solvent was removed in vacuo to yield the desired diiodo 5b as a brown solid (1.23 g, 98 %).
-
31Tetrahedron, 2005
-
32J. Acta Crystallogr. E: Crystallogr. Commun, 2015
mw = 466.10 g/mol. mp = 101 °C. IR (KBr, cm−1): ν = 3043, 2929, 1557, 1470, 1437, 1206, 1151, 1030, 755. 1H-RMN (400 MHz, CDCl3, ppm) δ =7.45 (dd, 3 J = 7.56 Hz, 4 J = 1.51 Hz, 2H, H7), 7.22 (td, 3 J = 7.38 Hz, 4 J = 1.55 Hz, 2H, H6), 7.17 (td, 3 J = 7.49 Hz, 4 J = 1.56 Hz, 2H, H5), 7.11 (dd, 3 J = 7.70 Hz, 4 J =1.47 Hz, 2H, H4), 4.64 (s, 4H, H1). 13C{1H}-RMN (100 MHz, CDCl3, ppm) δ = 139.9 (C3), 134.3 (C2), 133.1 (C4), 130.7 (C7), 129.2 (C5), 128.3 (C6), 4.11 (C1).
Results and discussion
Synthesis
The overall linear synthesis of the respective diol compounds Ch(C6H4CH2 OH)2 were based on a regioselective double-lithiation of the corresponding diphenyl(thio)ether, followed by a formylation reaction with DMF in situ; then a reduction of the resulting -CHO groups was carried out. Afterwards, the synthesis of the dichloro compounds 3a and 3b was carried out under Appel conditions, by using carbon tetrachloride and triphenylphosphine [33,34]. Dibrominated compounds 4a and 4b were obtained from a substitution reaction of the hydroxyl groups, by using Br− as a nucleophile. Finally, the Br-by-I halide exchange to obtain 5a and 5b was achieved with an excess of potassium iodide in acetone, by using 4a and 4b as starting materials.
-
33Angew. Chem. Int. Ed. Engl, 1975
-
34J. Org. Chem, 1978
NMR analysis
NMR spectra of all compounds were recorded in CDCl3 solutions at 25 °C; the assignment of the signals was carried out by the heteronuclear and homonuclear correlation of two-dimensional experiments (COSY, HSQC and HMBC). The signals of the aromatic protons of the ligand framework were observed as an ABCD pattern. In 13C-NMR, six signals are observed for the different aromatic carbons; the signal found in higher frequency correspond to the carbons directly bonded to the heteroatom, due to the electron-withdrawing effect. This effect is more noticeable when Ch = O due to its higher electronegativity compared to the sulfur atom. On the other hand, the -CH2- signals for both series of compounds (Ch = O, S) were used to monitoring that the chemical transformations were successful. The changes of these signals are clearly noticeable on 13C-NMR. Thus, when the carbon atom corresponds to the CHO functional group, the signal is around 190 ppm, and when the reduction reaction is carried out for its transformation into the -CH2-OH group, the carbon signal shifts ca. 60 ppm low-frequency. Then, the carbon resonances for -CH2-X were observed at 41.2, 28.2, 44.5, and 31.8 ppm for 3a, 4a, 3b, and 4b, respectively; when the halogen exchange occurs to obtain -CH2-I, a considerable change in the displacement of these carbons towards frequencies close to 0 ppm was observed, this due to the electro-donating effect exerted by the iodine atom.
X-Ray single crystal analysis Molecular structures of Ch(C6H4CH2X)2
There are some general features in the structural data of the 3a-5a diphenylether and 4b and 5b diphenylthioether corresponding sets. In all compounds, the Csp2-Ch and Csp3-X bond distances are comparable to the covalent radii sum [Ʃrcov(Csp2, O) = 1.39 Å; Ʃrcov(C sp2, S) = 1.78 Å; Ʃrcov(C sp3, Cl) = 1.78 Å; Ʃrcov(C sp3, Br) = 1.96 Å; Ʃrcov(C sp3, I) = 2.15 Å][35]. Also, the C-Ch-C angles are practically equal in each set, being the C-O-C angle the wider one. The difference in C-Ch-C angles is the expected; this bond angle decreases as the atomic number of the chalcogen atom (Ch) increases, as has been discussed by Prof. Gillespie for several compounds of the p -block, where the central atom has unshared lone pairs. [36] To analyze the conformations of the compounds in each set, we overlayed each pair of molecules (Figures 3 and 4). We used the Mercury 4.0 software [37] to calculate the root mean square deviation (RMSD) and the maximum deviation data in these superimpositions. Thus, we observed striking differences in the conformation of the diphenylether derivatives 3a-5a in the solid state, despite the similitude of their skeletal structures. Firstly, the conformations of 3a and 4a are very similar (RMSD = 0.154 Å), regardless of their different crystalline systems and space groups (vide infra). The main conformational difference of this pair in comparison with 5a lays on the direction of the C-X bonds; in 3a (X = Cl) and 4a (X = Br), the halogen atoms are pointed to opposite sides, while in the diiodo compound, the C-I bonds are oriented to the same side. For the diphenylthioethers 3b and 4b, the conformations are essentially the same (RMSD = 0.064 Å, with a maximum deviation of 0.154 Å, mainly due to the expected difference in C-X bond lengths). In these compounds, the halogen atoms are also pointed to opposite sides. We also analyzed the C2-C3-Ch-C10 and C9-C10-Ch-C3 torsion angles. As a result, excepting the data for 5a, the difference of these angles (|Δta|) is lesser than 10.3°, indicating a practically two-fold rotational symmetry (in fact, 4a displays a perfect C2 axis that bisects the C-O-C bond angle, with an obviously null difference in torsion angles). On the other hand, 5a shows a difference of 76.7° that is consistent with an almost orthogonal arrangement of both phenyl rings around the C-O-C plane.
-
36Chemical Bonding and Molecular Geometry; From Lewis to Electron densities, 2001
-
37J Appl. Cryst, 2020
Crystal arrangement of Ch(C6H4CH2X)2 derivatives
The chloro derivative 3a crystallized in the triclinic system, while 4a and 5a pair crystallized in the monoclinic system, but in different space groups. On the other hand, both diphenylthioether compounds 4b and 5b, besides to be isostructural, they are also isomorphic, crystallizing in the centrosymmetric triclinic system, with a higher cell volume in 5b due to the larger size of the iodine atoms. In all Ch(C6H4CH2 X)2 derivatives, notwithstanding the different nature of either Ch or X atoms, were observed non-classical C-H···X hydrogen bonding as well as p-interactions that contributes to the cohesion of the crystal (Fig. 5).
Thumbnail
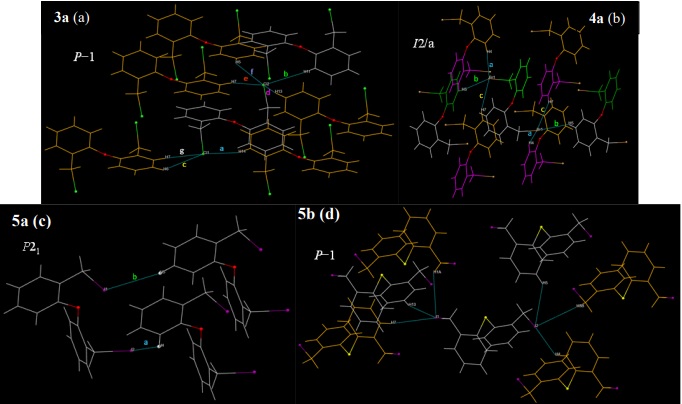
Fig. 5
C-H···X hydrogen bonding in Ch(C6H4CH2 X)2 derivatives (colours are used to highlight the symmetry operation involved in the generation of symmetry-related moieties; see text).
C-H···X hydrogen bonding in Ch(C6H4CH2 X)2 derivatives (colours are used to highlight the symmetry operation involved in the generation of symmetry-related moieties; see text).
In the Fig. 5 we plotted a portion of the crystal structure where the molecules were related by their symmetry operators and, for the sake of comparison, we highlighted the C-H···X hydrogen bonding around the X atom in the asymmetric unit. In all plots, the asymmetric unit is displayed in gray color; the other colors are used for moieties related by other symmetry operation, such as inversion (in yellow) or, in the case of 4a, rotation, rotation-inversion (both in green), and gliding (in magenta). The tagged distances of the intermolecular contacts and the symmetry operators for all compounds are summarized in Table 3. For 4a, the asymmetric unit is just the half, with the oxygen atom positioned on a two-fold axis; the other half is generated by the corresponding two-fold rotation. As commented, 4b and 5b were isomorphic and, for conciseness, we just graph the structure of the iodo derivative. To build all the plots showed, we considered intermolecular H···X contacts smaller than 3.6 Å. From the plots, the number of contacts in the diphenylether derivatives decreases in the order 3a>4a>5a; the magnitude of the distances must also decrease but in the inverse order because of the different atom sizes. On the other hand, the number of intermolecular interactions is the same in the diphenylthioethers 4b and 5b, as can be expected, because of their isomorphism.
Table 3
Structural data of C-H···X hydrogen bonding in Ch(C6H4CH2 X)2 derivatives and symmetry operators.
Structural data of C-H···X hydrogen bonding in Ch(C6H4CH2 X)2 derivatives and symmetry operators.
| tag | 3a | 4a | 5a | 4b | 5b |
| a | 2.918; C–H14···Cl1 1−x, 1−y, 1−z | 3.047; C–H4···Br1 x, 1/2−y, −1/2+z | 3.358; C–H4···I2 −1+x, y, z | 3.160; C–H4···Br2 −1+x, y, z | 3.239; C–H4···I2 1−x, 1−y, 1−z |
| b | 3.106; C–H11···Cl2 −1+x, y, z | 3.186; C–H5···Br1 −1/2+x, −1/2+y, −1/2+z | 3.371; C–H5···I1 −1+x, 1+y, z | 3.302; C–H13···Br1 x, y, −1+z | 3.329; C–H13···I1 x, y, −1+z |
| c | 3.108; C–H6···Cl1 2−x, 1−y, 2−z | 3.207; C–H7···Br1 −1/2+x, 1−y, z | Not applicable | 3.333; C–H5···Br2 x, y, −1+z | 3.372; C–H5···I2 −1+x, y, z |
| d | 3.130; C–H13···Cl2 1−x, −y, 1−z | Not applicable | Not applicable | 3.374; C–H7···Br1 2−x, 2−y, −z | 3.574; C–H7···I1 2−x, 2−y, −z |
| e | 3.132; C–H7···Cl2 1−x, 1−y, 2−z | Not applicable | Not applicable | 3.454; C–H8B···Br2 −x, 1−y, 1−z | 3.582; C–H8B···I2 −x, 1−y, 1−z |
| f | 3.181; C–H5···Cl2 1−x, −y, 2−z | Not applicable | Not applicable | 3.469; C–H1A···Br1 1−x, 2−y, −z | 3.499; C–H1A···I1 1−x, 2−y, −z |
| g | 3.254; C–H7···Cl1 2−x, 1−y, 2−z | Not applicable | Not applicable | Not applicable | Not applicable |
Hirshfeld surface analysis
The Hirshfeld surface (HS) encloses either a molecule or a cluster of molecules, and its corresponding 2D-fingerprint is unique for a given compound, therefore, the construction of this type of surfaces makes them a useful approach to compare crystal structures [23]. The surfaces based on the d norm of all Ch(C6H4CH2 X)2 compounds clearly exhibit red spots in the vicinity of the X substituents, corresponding to the presence of close contacts due to C-H···X non-classical hydrogen bonding, i.e., contact distances that are shorter than the van der Waals radii sum [3a(1), 4a(1), 5a(1), 4b(1), and 5b(1) plots] in Fig. 6. The fragment patch plots are useful to calculate the number of molecules that interacts with a central one; also, the area data of these patches can be used to find the external major molecular fragments that are closer to a HS given. In Fig. 6 are plotted the corresponding fragment patches over the HS [3a(2), 4a(2), 5a(2), 4b(2), and 5b(2) plots]. In the O(C6H4CH2 X)2 compounds, the number of interacting molecules is 15, 20, and 16, for 3a, 4a, and 5a, respectively (we considered a radius of 4.10 Å as an atom---atom distance, instead of the default value of 3.80 Å in CrystalExplorer). The four larger areas over the HS surface are 52.5, 43.0, 35.6, and 21.7 Å2 for 3a (52.5 % superficial area); 52.7, 51.6, 42.0, and 42.0 Å2 for 4a (63.0 % superficial area); and 63.6, 61.5, 29.9, and 20.3 Å2 for 5a (57.0 % superficial area). For the isomorphic 4b and 5b there are 16 molecules around the HS in each one; the larger areas are 52.5, 47.8, 44.5, and 44.1 Å2 for 4b (62.2 % superficial area); and 55.0, 48.4, 47.3, and 43.5 Å2 for 5b (61.7 % superficial area). Overall, four interacting molecules cover more than 52 % of each corresponding HS, despite their different surrounding number (see above).
-
23J Appl. Cryst, 2021
Thumbnail
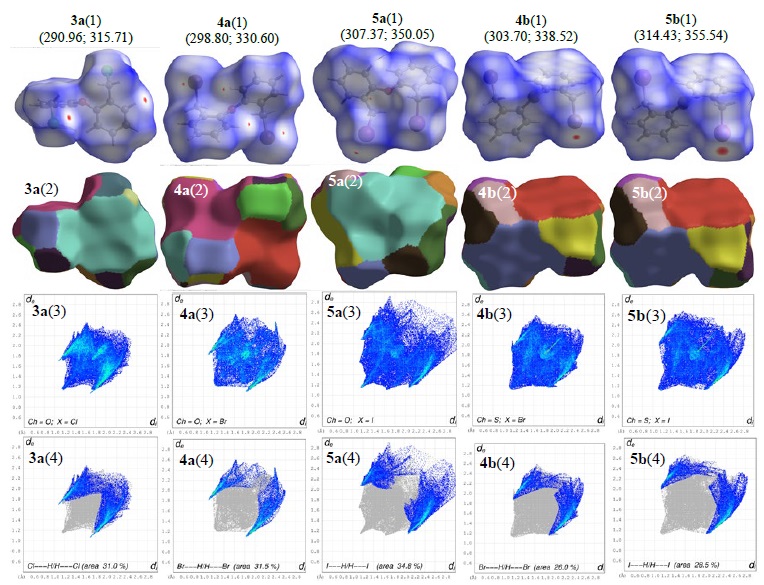
Fig. 6
Hirshfeld surfaces mapped over d norm, fragment patches, and fingerprint plots for Ch(C6H4CH2 X)2 (Ch = O, S; X = Cl, Br, I) compounds (data in parentheses are superficial area in Å2 and volume in Å3).
Hirshfeld surfaces mapped over d norm, fragment patches, and fingerprint plots for Ch(C6H4CH2 X)2 (Ch = O, S; X = Cl, Br, I) compounds (data in parentheses are superficial area in Å2 and volume in Å3).
To evaluate the contribution of the intermolecular contacts we plotted the 2D-fingerprints of the corresponding Hirshfeld surfaces [3a(3), 4a(3), 5a(3), 4b(3), and 5b(3) plots]. For the sake of comparison, we decomposed the corresponding overall 2D fingerprints plots in their X···H/H···X [X = Cl, Br, I; 3a(4), 4a(4), 5a(4), 4b(4), and 5b(4) plots in Fig. 7] fingerprint plots for all compounds. We also plot the contribution of all contacts by means of a stacked bar chart, where we consider the reciprocal ones. The analysis of the intermolecular contact contributions for all compounds showed that H···H contacts are dominant, from 43.6 to 37.7 % contribution (Fig. 7). The next predominant non-covalent contributions are the X···H/H···X non-classic hydrogen bonding (green bars); we observed that compounds with Ch = O display higher values, probably due to the higher electronegativity of the oxygen atom in 3a-5a when compared to the sulfur atom in 4b and 5b. To finish, the C···H/H···C (black bars) also display significant contributions due to the presence of the aromatic rings, which ones enhance C-H···pi as well as pi···pi interactions.
Thumbnail
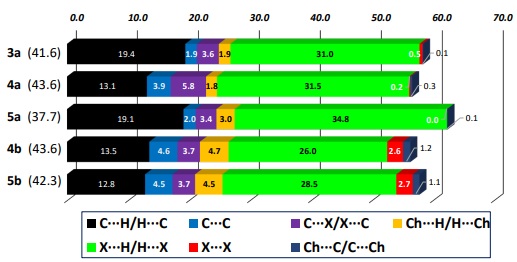
Fig. 7
Relative contributions in percentage of various intermolecular contacts of Ch(C6H4CH2 X)2 (Ch = O, S; X = Cl, Br, I) molecular crystals (H···H contacts are in parentheses).
Relative contributions in percentage of various intermolecular contacts of Ch(C6H4CH2 X)2 (Ch = O, S; X = Cl, Br, I) molecular crystals (H···H contacts are in parentheses).
A deeper analysis indicated that 4a displayed a high C···Br/Br···C contribution (5.8 %, purple bar) in a centrosymmetric arrangement, and the isomorphic 4b/5b pair displayed X···X notable contributions (red bars). In the first case, 4a displayed C-Br···pi interactions alongside with other two C-H···Br interactions; the distance from this bromo atom to the centroid of the three C4, C5, and C6 aromatic atoms is 3.801 Å that is longer than the van der Waals radii sum [ƩrvdW(C, Br) = 3.55 Å]. The analysis of their Hirshfeld surfaces decorated with the molecular electrostatic potential (MEP) showed that the centrosymmetric arrangement of two molecules effectively displayed complementary interactions (see Fig. 8). The bromo atom in the C-Br bond display at its σ-hole position a positive potential (5.96 kcal/mol, in blue colour), while the three C(4), C(5), and C(6) aromatic atoms display a complementary negative potential (7.03 kcal/mol, in red colour) that is indicative of a C-Br···pi interaction of attractive nature. Something similar is observed for the C-H(4)···Br interaction, where the negative belt around the Br atom is displaying a negative potential (−15.06 kcal/mol, in red colour); as expected, the hydrogen atom in the C-H(4) bond has a positive potential (14.62 kcal/mol, in blue colour).
Thumbnail
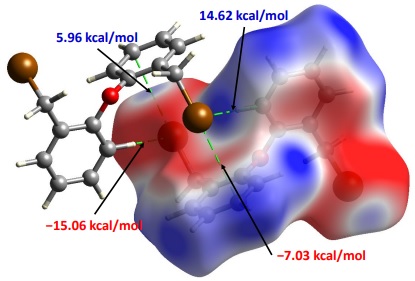
Fig. 8
Views of calculated electrostatic potential mapped on the Hirshfeld surface of 4a showing the potential at the points of contact for Br···pi and C-H···Br noncovalent interactions.
Views of calculated electrostatic potential mapped on the Hirshfeld surface of 4a showing the potential at the points of contact for Br···pi and C-H···Br noncovalent interactions.
Finally, as has been commented, the isomorphic 4b/5b pair displayed X···X interactions (X = Br, I). In Fig. 9 we show the corresponding Hirshfeld surfaces decorated with the MEP to highlight just these interactions. In the front and rear plots, it is worth mentioning that there are differences in the positions of the head black arrow and the dashed lines that specify the shortest X···X interatomic distances intersecting the HS decorated with the MEP. These positions are different because of we took the potential energy data from the intersection of the surface with the straight line coming from either outer or inner C-X bond, since in this position is where the s-hole is located. Each compound displays both complementary positive and negative potentials; the C-Br(1) and C-I(1) inner bonds display positive potentials of 7.53 and 11.61 kcal/mol for 4b and 5b, respectively (in blue color) while the matching negative potentials are −8.22 and −11.23 kcal/mol (in red color). Also, in the rear plots we show other positive potentials of 7.65 and 15.19 kcal/mol that match with the C(12) and C(13) benzenic carbons, similar to that observed in 4a.
Thumbnail
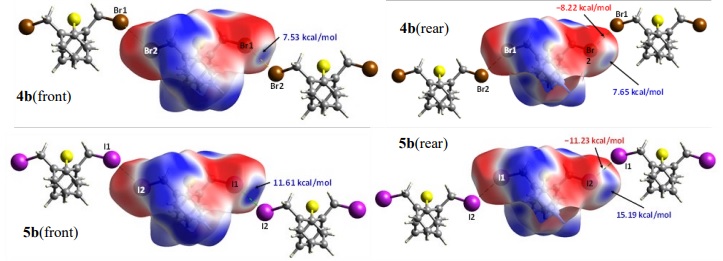
Fig. 9
Views of calculated electrostatic potential mapped on the Hirshfeld surface of 4b and 5b showing the potential at the points of contact for C-X···X-C (X = Br, I) noncovalent interactions.
Views of calculated electrostatic potential mapped on the Hirshfeld surface of 4b and 5b showing the potential at the points of contact for C-X···X-C (X = Br, I) noncovalent interactions.
Conclusions
Two series of dihalogenated compounds of general formula Ch(C6H4CH2 X)2 were synthetized by a linear approach from either cheap diphenylether or diphenylthioether used as starting materials. Despite the similitude in the skeletal structure, all compounds based on the ether framework displayed different interactions in the crystal arrangement. On the other hand, the thioether compounds were isostructural and isomorphic and, in these two cases we observed that the bromo- and iodo compounds were more susceptible to form X···X interactions and displayed little disposition to form X···H hydrogen bonding, due to the presence of the less electronegative sulfur atom as compared with the oxygen ones in the diphenylethers.
Acknowledgements
JVGG fully acknowledges the scholarship from CONACYT (ID 791450). This research was supported by project A1‐S‐12381 (CONACYT). CGGP acknowledges the financial assistant support from the same project (ID DIDI-DI- BECA-001).
References
- 1. Dean, J. A., in: Lange's Handbook of Chemistry (15th Edition), McGraw-Hill Education, New York, 1999, 330. Links
- 2. Häggblom, M. M.; Bossert, I. D., in: Halogenated Organic Compounds - A Global Perspective. In Dehalogenation. Springer, Boston, MA, 2004, 3-29. DOI: https://doi.org/10.1007/0-306-48011-5_1. Links
- 3. Patai S., Rappoport Z., in: Halides, Pseudo‐Halides and Azides; Part 1 and Part 2 in The Chemistry of Functional Groups, John Wiley & Sons Ltd, 1983, 1-223. DOI:10.1002/9780470771716. Links
- 4. Anderson B. M., Meyers R. A.; in: Halogen Chemistry. In Encyclopedia of Physical Science and Technology (Third Edition), Academic Press, Elsevier, 2003, 197-222. DOI: https://doi.org/10.1016/B0-12-227410-5/00307-0. Links
- 5. Smolnikov, S.; Bin Shahari, M.; Dolzhenko, in: Green sustainable process for chemical and environmental engineering and science: A. Sonochemical protocols for Grignard reactions. Elsevier, 2020, 243-255. DOI: 10.1016/B978-0-12-819540-6.00009-7. Links
- 6. Odd Hassel - Nobel Lecture. NobelPrize.org. Nobel Prize Outreach AB 2023. Sun. 19 Mar 2023. https://www.nobelprize.org/prizes/chemistry/1969/hassel/lecture/ Links
- 7. Desiraju, G. R.; Ho, P. S.; Kloo, L. Legon, A. C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Pure Appl. Chem. 2013, 85, 1711-1713. DOI: http://dx.doi.org/10.1351/PAC-REC-12-05-10. Links
- 8. Cavallo, G.; Metrangolo, P.; Milani, R.: Pilati, T.; Priimagi, A.; Resnati, G. Terraneo, G. Chem. Rev. 2016, 116, 2478-2601. DOI: https://pubs.acs.org/doi/10.1021/acs.chemrev.5b00484. Links
- 9. Metrangolo, P .; Neukirch, H. Pilati, T .; Resnati, G . J. Am. Chem. Soc. 2005, 38, 386-395. DOI: https://doi.org/10.1021/ar0400995. Links
- 10. Sutar, R. L.; Huber, S. M. J. Am. Chem. Soc . 2019, 9, 9622-9639. DOI: https://doi.org/10.1021/acscatal.9b02894. Links
- 11. Ding, X., Tuikka, M.; Haukka, M., in: Halogen Bonding in Crystal Engineering: Recent Advances in Crystallography, IntechOpen, 2012, 143-168. DOI: https://doi.org/10.5772/48592. Links
- 12. Nunzi, F.; Cesario, D.; Tarantelli, F.; Belpassi, L. Chem. Phys. Lett. 2021, 771, 138522. DOI: https://doi.org/10.1016/j.cplett.2021.138522. Links
- 13. Prasanna, M.D.; Guru Row, T.N. Cryst. Eng. 2000, 3, 135-154. DOI: https://doi.org/10.1016/S1463-0184(00)00035-6. Links
- 14. Zhao-Qi, G.; Shi-Hui, Q.; Huan-Hui, Y.; Shan-Chao, W.; Meng, Z.; Gui-Mei, T.; Yong-Tao, W.; Tao, A.; Seik-Weng N. J. Mol. Struct. 2022, 1267, 133606. DOI: https://doi.org/10.1016/j.molstruc.2022.133606. Links
- 15. Mejia-Rivera, F. J.; Alvarado-Rodríguez, J. G.; Andrade-López, N.; Cruz-Borbolla, J.; Jancik, V. Inorg. Chem. Commun. 2018, 97, 44-48. DOI: https://doi.org/10.1016/j.inoche.2018.09.006. Links
- 16. Nather, C.; Jess, I.; Kus, P.; Jones, P.G. Cryst. Eng Comm. 2016, 18, 3142 Links
- 17. Xu, X.; Strongin, R.M.; Fronczek, F.R. CSD Communication (Private Communication), 2015. Links
- 18. Oxford Diffraction CrysAlis software system, version 1.171.37.35. Oxford Diffraction Ltd., Abingdon, UK (2014) Links
- 19. Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann H. J. Appl. Cryst. 2009, 42, 339-341. Links
- 20. Sheldrick, G.M. Acta Cryst. A. 2015, 71, 3-8. Links
- 21. Sheldrick, G.M. Acta Cryst. C. 2015, 71, 3-8. Links
- 22. Spackman, M.A.; Jayatilaka, D. CrystEngComm. 2009, 11, 19-32 Links
- 23. Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. J Appl. Cryst . 2021, 54,1006-1011. Links
- 24. Jayatilaka, D.; Grimwood D.J. Comput. Sci. ICCS, 2003, 4, 142-151. Links
- 25. Becke A.D. J. Chem. Phys. 1993, 98, 5648-5652 Links
- 26. Godbout, N.; Salahub, D. R.; Andzelm J.; Wimmer Can, E. J. Chem. 1992, 70, 560-571. Links
- 27. Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon D.A. J. Phys. Chem. 1992, 96, 6630-6636. Links
- 28. Osuka, A.; Kobayashi, F.; Maruyama, K. Bull. Chem. Soc. Jpn. 1991, 64, 1213-1225. DOI: https://doi.org/10.1246/bcsj.64.1213. Links
- 29. Britovsek, G. J. P.; Gibson, V. C.; Hoarau, O. D.; Spitzmesser, S. K.; White, A. J. P.; Williams, D. J. Inorg. Chem. 2003, 42, 3454-3465. DOI: https://doi.org/10.1021/ic034040. Links
- 30. Martínez-Otero, D.; Alvarado-Rodríguez, J. G.; Cruz-Borbolla, J.; Andrade-López, N.; Pandiyan, T.; Moreno-Esparza, R.; Flores-Alamo, M.; Cantillo-Castillo J. Polyhedron. 2012, 33, 367-377. DOI: https://doi.org/10.1016/j.poly.2011.11.053. Links
- 31. Kida, T.; Kikuzawa, A.; Higashimoto, H.; Nakatsuji, Y.; Akashi, M. Tetrahedron. 2005, 61, 5763-5768. DOI: https://doi.org/10.1016/j.tet.2005.04.026. Links
- 32. McAdam, C.J.; Hanton, L.R.; Moratti, S.C.; Simpson, J. Acta Crystallogr. E: Crystallogr. Commun. 2015, 71, 1505-1509. DOI: 10.1107/S2056989015021295. Links
- 33. Appel, R. Angew. Chem. Int. Ed. Engl. 1975, 14, 801-811. DOI: https://doi.org/10.1002/anie.197508011. Links
- 34. Cristol, S. J.; Strom, R. M.; Stull, D. P., J. Org. Chem. 1978, 43, 1150. DOI: https://doi.org/10.1021/jo00400a027. Links
- 35. Cordero, B.; Gómez, V.; Platero-Prats, A. E.; Revés, M.; J. Echeverría, J., Cremades, E.; Barragán F.; Alvarez, S. Dalton Trans. 2008, 21, 2832-2838. DOI: https://doi.org/10.1039/B801115J. Links
- 36. Gillespie, R.J. Popelier, P.L.A., in: Chemical Bonding and Molecular Geometry; From Lewis to Electron densities. Oxford University Press, New York, 2001, 84. DOI: 10.1021/ed080p31. Links
- 37. Macrae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A. J Appl. Cryst . 2020, 53, 226-236. DOI: https://scripts.iucr.org/cgi-bin/paper?gj5232. Links