
De la Secuenciación a la Aceleración Hardware de los Programas de Alineación de ADN, una Revisión Integral
From sequencing to hardware acceleration of DNA alignment software: A integral review
D. Pacheco Bautista1, M. González Pérez1, I. Algredo Badillo2
Universidad Popular Autónoma del Estado de Puebla.1
]]> Universidad Politécnica de Tlaxcala.2
Correspondencia:
Daniel Pacheco Bautista
Ciudad Universitaria S/N Barrio Sta. Cruz Tagolaba, C.P. 70760, Tehuantepec, Oaxaca.
Correo electrónico: dpachecob@bianni.unistmo.edu.mx
Fecha de recepción: 8 de julio de 2015.
Fecha de aceptación: 3 de septiembre de 2015.
]]> RESUMEN
En los últimos años ha ocurrido un avance impresionante en las máquinas de secuenciación paralela masiva, también llamadas de secuenciación de siguiente generación (NGS), por ejemplo, máquinas recientes como Illumina Hiseq son capaces de generar millones de lecturas en una sola corrida. No obstante, estas tecnologías están limitadas a secuenciar solo fragmentos pequeños de material genético (entre 35 y 1100 nucleótidos), por lo que para secuenciar un genoma completo es necesario dividir la cadena, secuenciar y posteriormente ensamblar las lecturas cortas obtenidas. En este trabajo se revisan y comparan las tecnologías de secuenciación recientes, se estudia el proceso de ensamble de genomas completos y se establece formalmente el problema de la alineación. También se incluye un resumen de los principales programas de alineación y sus algoritmos que lo soportan. Finalmente, después de concluir que las tecnologías de secuenciación han superado en velocidad por un factor mayor a 10x a los programas de alineación, se revisa la aceleración Hardware como alternativa para acelerar tales programas. Este trabajo al ser una revisión integral pretende contribuir al desarrollo de investigación en el área de bioinformática en el país.
Palabras clave: secuenciación NGS, mapeo, bioinformática, aceleración hardware.
ABSTRACT
In recent years, impressive progress has occurred in the machines of massively parallel sequencing, also called of next-generation sequencing (NGS), for example, recent machines like Illumina HiSeq are capable of generating millions of reads in a single run. However, these technologies are limited to sequence only small fragments of genetic material (35 to 1100 nucleotides), so that for complete-genome sequencing, it is necessary to divide the chain, to sequence the fragments, and, subsequently, to assemble the obtained short readings. In this paper, the recent NGS sequencing technologies are reviewed and compared, analyzing the problem of sequence assembly, and formally establishing the problem of alignment. Also, it is examined the main alignment programs and the algorithms that support them. Finally, after concluding that sequencing technologies have speed that exceeds 10 times to the speed of the alignment programs, the hardware acceleration is reviewed as an alternative to accelerate these programs. This work, which is a comprehensive analysis and review, aims to contribute to the development of the research in the area of bioinformatics in the country.
Keywords: NGS sequencing, mapping, bioinformatics, hardware acceleration.
INTRODUCCIÓN
La secuenciación de ADN, es el proceso mediante el cual se determina el orden de las bases nucleicas (Adenina, Guanina, Citosina y Timina) dentro de una cadena de ADN, siendo una tarea clave en biología molecular, genómica y medicina (1). En los últimos años, los avances tecnológicos han hecho posible secuenciar en forma más económica y rápida enormes cantidades de material genético abriendo oportunidades sin precedentes para la investigación y el diagnostico. La secuenciación de ADN tiene una lista larga y versátil de aplicaciones, siendo una tecnología clave en la investigación de algunos tipos de cáncer, así como del VIH, ayuda también a incrementar el conocimiento básico de organismos y células, diagnostico de pacientes, investigación a la resistencia de drogas, o predisposición a enfermedades. Adicionalmente el precio de secuenciar un genoma humano pronto caerá por debajo de 1000 dólares, este precio ha sido considerado por mucho tiempo el punto clave para la generalización de la medicina personalizada. La idea es que una vez que el precio caiga por debajo de esa cantidad, finalmente sea suficientemente efectivo en costo para permitir a los médicos, entregar tratamientos basados en la genética del paciente. En lugar de administrar tratamientos basados en exámenes y síntomas, el médico será capaz de revisar el genoma del paciente para diagnosticar y perfeccionar tratamientos desde la diabetes hasta el alzhéimer. Lamentablemente, las máquinas de secuenciación actuales están limitadas a secuenciar segmentos cortos de ADN, por lo que para secuenciar un genoma completo es necesario dividirlo en segmentos, secuenciarlos y posteriormente ensamblar los códigos obtenidos mediante software sofisticado con elevada complejidad temporal y espacial. Estos programas aun cuando tiene soporte para ejecución en múltiples núcleos y/o múltiples procesadores, no pueden competir con la velocidad de las máquinas de secuenciación modernas, representando el cuello de botella del proceso de análisis. En este artículo se presenta un panorama global de la secuenciación enfatizando en el problema de la alineación, con un estudio de los programas más utilizados y los algoritmos que los soportan, así como una descripción de las posibles alternativas para su aceleración mediante hardware especializado.
]]>SECUENCIACIÓN Y TECNOLOGÍAS NGS
La secuenciación de ADN tiene sus inicios en 1977, cuando Frederick Sanger y equipo desarrollaron el método de secuenciación enzimática, también conocida como secuenciación Sanger, didesoxi o de finalización de cadena (2), y Maxam y Gilbert desarrollaron el método de secuenciación química (3). Debido a su alta eficiencia y baja radioactividad, la secuenciación Sanger fue adoptada como la tecnología primaria en la "primer generación" de aplicaciones de secuenciación comerciales y de laboratorio. En sus inicios, la secuenciación Sanger era laboriosa y requería de materiales radioactivos, después de años de mejora Applied Biosystems introdujo la primera máquina de secuenciación automática (nombrada ABI370) en 1987, adoptando electroforesis capilar (EC), la cual hizo el proceso más rápido y exacto. AB370 podía detectar 96 nucleótidos simultáneamente, 500K nucleótidos al día, y las longitudes de lectura alcanzaban los 600 nucleótidos. Los instrumentos de secuenciación automática basados en EC y secuenciación Sanger, así como el software asociado, llegaron a ser las herramientas principales para la culminación del proyecto del genoma humano en el 2003 (4). Este proyecto estimuló fuertemente el desarrollo de nuevos instrumentos de secuenciación para incrementar la velocidad y exactitud reduciendo simultáneamente los costos y la mano de obra, surgiendo, en el 2005, las tecnologías de secuenciación nombradas de siguiente generación o tecnologías NGS, las cuales difieren del método de Sanger fundamentalmente en el uso exhaustivo de tecnología paralela. En el 2005, 454 Life Sciences lanza al mercado el secuenciador 454, un año después Solexa hace lo propio con el secuenciador Genome Analyzer (Que recientemente evolucionó a Hiseq), seguido por el lanzamiento de SOLID de la empresa Agencourt, CGA de complete Genomics y PacBio RS de Pacific Biosciences, los cuales son sistemas representativos de secuenciación paralela masiva o tecnología NGS. Algunas de estas compañías fundadoras fueron compradas posteriormente por otras compañías: En el 2006 Agencourt fue comprada por Applied Biosystem, y en el 2007, 454 fue comprada por Roche, mientras que Solexa fue comprada por Ilumina. Después de años de evolución los sistemas NGS exhiben cada vez mejor desarrollo y ventajas propias de la tecnología específica, como se muestra en la Tabla 1. En general algunas tecnologías ya superan el Terabyte de lecturas producidas por corrida, mientras que otras alcanzan exactitudes comparables con la lograda por el método de Sanger. No obstante la longitud de lecturas limitada a valores iguales o inferiores a 1100 nucleótidos sigue siendo su principal limitante. Actualmente los secuenciadores NGS son comercializados por un número importante de empresas, cada una desarrollando y aplicando diferentes métodos y tecnologías (Tabla 2), sin embargo a pesar del dinamismo tecnológico hay principios generales utilizados en la construcción de tales dispositivos.
Las plataformas NGS comparten tres pasos fundamentales: Preparación de la muestra, inmovilización y detección (Figura 1) (5) (6) (7). Generalmente la preparación de la muestra, involucra la adición de secuencias de ADN comunes o universales, conocidas como "adaptadores", a los extremos de hebras de ADN fragmentado aleatoriamente, la preparación resultante es nombrada "librería de secuenciación". En la etapa de inmovilización, los adaptadores se utilizan para sujetar los fragmentos de ADN a una superficie sólida, de esta manera definiendo el sitio en el cual la reacción de secuenciación comenzará, adicionalmente, de secuenciación se amplifica para formar a excepción de PacBio RS, la librería características de secuenciación detectables y distinguidas espacialmente. El paso final del proceso es la detección. Las plataformas de secuenciación NGS integran una variedad de tecnologías ópticas y de fluidos, para desarrollar y monitorear las reacciones de secuenciación molecular, las cuales pueden ser mediante síntesis de la polimerasa de ADN o ligación de oligonucleótidos fluorescentes. Cada ciclo de detección consiste en la incorporación de un sustrato de ácido nucleico detectable al templado inmovilizado, lavado y captura de imágenes o señales del evento molecular mediante sistemas ópticos de alta velocidad. El ciclo de incorporación, lavado y captura se repite hasta obtener la lectura de la secuencia completa de ADN.
SECUENCIACIÓN DE CADENAS LARGAS DE ADN
Las lecturas obtenidas de los instrumentos de secuenciación de cualquier generación son demasiado cortas como para cubrir regiones de interés en investigación genómica, por lo que fue necesario el desarrollo de métodos que permitieran secuenciar segmentos más largos de ADN e incluso de genomas completos (WGS, del inglés Whole Genome Sequencing). La estrategia más utilizada se conoce con el nombre de secuenciación Shotgun (8), y se ilustra de manera simplificada en la Figura 2. En esta técnica la cadena de ADN a secuenciar se clona a través del uso de PCR o mediante una bacteria anfitrión, el número de copias que se obtienen se conoce como cobertura. Posteriormente la muestra resultante se divide aleatoriamente en pequeños fragmentos y se secuencía en forma desordenada mediante alguna de las tecnologías revisadas previamente, la división aleatoria crea fragmentos de diferente tamaño por lo que en este paso es necesario elegir únicamente las que se encuentran en un rango apropiado para la tecnología de secuenciación a utilizar. Una vez obtenidas las lecturas, los traslapes entre estas se utilizan para reconstruir mediante técnicas computacionales sofisticadas la secuencia original, a este último paso se le conoce como ensamble de fragmentos.
El ensamble de fragmentos es una tarea ardua, realizada mediante técnicas computacionales de elevadas prestaciones. El objetivo es reconstruir la cadena que representa el código genético de la molécula original a partir de los millones de lecturas obtenidas mediante las máquinas NGS. Lo anterior puede llevarse a cabo mediante dos formas, en la primera la reconstrucción se realiza utilizando como referencia un genoma secuenciado previamente, tal mecanismo recibe el nombre de alineación o mapeo. La segunda reconstruye la cadena a partir exclusivamente de las lecturas secuenciadas, y es conocida como ensamble De Novo o simplemente Ensamble. El resto de este artículo está dedicado a la alineación o mapeo, referimos al lector interesado en ensamble De Novo a consultar las referencias (10) y (11) para un estudio más profundo.
ALINEACIÓN DE LECTURAS CORTAS
]]> La alineación de lecturas cortas se utiliza en proyectos de re-secuenciación, donde se obtiene el código genético de miembros de una especie que ha sido secuenciado previamente mediante un método De Novo. El ejemplo más clásico es el del ser humano, en tales proyectos se persigue encontrar variaciones genómicas que caractericen en forma particular a un individuo mediante comparación con los resultados obtenidos del proyecto del genoma humano, intentando explicar tópicos como susceptibilidad a enfermedades, rasgos fisiológicos característicos, resistencia a drogas, etc. En tales aplicaciones, la elevada cantidad de datos generadas por las tecnologías NGS así como la limitada longitud de las lecturas producidas evita el uso de programas de alineación tradicionales como BLAST (12). Adicionalmente, a diferencia de las aplicaciones de alineación típicas, el interés ahora está centrado en localizar variaciones mínimas entre las lecturas y la referencia, es decir se procesan genomas que se espera tengan elevada similitud con el genoma de referencia. Para dimensionar el problema, en el ejemplo del genoma humano, el número de lecturas m es usualmente 107 - 108, la longitud de u na lectura l es de 35-1100 nucleótidos, la longitud del genoma |R| es 3 x 109 nucleótidos y las variaciones entre un humano y otro son apenas alrededor del 0.1% (13). Lo anterior ha incubado el desarrollo de nuevos programas de alineación de baja sensibilidad y alta velocidad denominados alineadores de lecturas cortas, cuyos principios de funcionamiento serán tratados en esta y en la siguiente sección.
Definición del problema de la alineación
A partir de la discusión previa y de la teoría de secunciación NGS, pueden determinarse algunas características importantes del problema de la alineación de lecturas cortas de ADN:
• El alfabeto está constituido por 4 letras Σ= {A, C, G, T} cada un a representando a un nucleótido, sin embargo cuando no se sabe el tipo de base en una secuencia es común usar el símbolo N en su lugar.
• El genoma de referencia es fijo y conocido previamente, su tamaño es del orden de unas decenas a miles de millones de nucleótidos. Lo que sugiere que pueda indexarse una sola vez y reutilizarse en cada proyecto de re-secuenciación de esa misma especie.
• Las lecturas tienen una longitud fija entre 35-1100 nucleótidos para las tecnologías de secuenciación actuales. La cantidad de lecturas suele ser muy grande del orden de millones por cada pasada de las máquinas NGS.
• La similitud entre el genoma de referencia y el genoma re-secuenciado es aproximadamente del 99.9 %.
Ante este panorama el problema del mapeo puede establecerse en forma general de la siguiente manera:
]]> Entradas:• Un conjunto de lecturas cortas de ADN f1,..., fm, cada una de longitud l.
• Una secuencia de referencia R de tamaño |R|
• Un grupo de restricciones del proceso
Salida:
• Posiciones en R en donde cada lectura se mapea exacta o aproximadamente.
El grupo de restricciones puede variar dependiendo de la plataforma específica de secuenciación NGS utilizada, así como del tipo de datos procesados, es decir, si los datos generados son lecturas sencillas o por pares, pero también de restricciones impuestas por el usuario. Es importante notar como la definición de mapeo establecida debe permitir alineaciones aproximadas, debido fundamentalmente a ciertas particularidades que ocurren en el genoma y en la secuenciación, las cuales serán revisadas a continuación.
Diferencias biológicas: Son diferencias locales pequeñas del genoma siendo considerado con respecto al genoma de referencia conocido para su especie, ocurren con una frecuencia de aproximadamente 1/1000 en el caso del genoma humano (13). Un ejemplo de estos son los polimorfismos de nucleótido simple (SNP), los cuales básicamente representan la sustitución de un solo nucleótido de la cadena de referencia (Figura 3a), los SNPs ocurren principalmente por la diversidad encontrada dentro de una misma especie, por ejemplo en el caso de los humanos un SNP podría ser responsable del color del cabello o la susceptibilidad a una enfermedad en particular. Otras variaciones biológicas que ocurren en secuencias genómicas son las inserciones y las supresiones (indels), en la cual un segmento de la lectura difiere de la referencia debido a la inserción o supresión de una o más bases. Los indels causan un desplazamiento de las bases con respecto al genoma de referencia, tal como se muestra en los incisos b y c de la Figura 3.
]]>
Errores de secuenciación: Un error de secuenciación ocurre cuando la máquina de secuenciación etiqueta incorrectamente una base. Por ejemplo en la Figura 4, se reporta un nucleótido de guanina (G) en el genoma, en donde un nucleótido de Adenina (A) ocurre realmente. Es posible diferenciarlo de un SNP debido a que un error máquina generalmente ocurre en una única fila de las lecturas, mientras que un SNP ocurre en todas las lecturas que cubren esa región. De esta forma la cobertura o profundidad de las lecturas cortas puede utilizarse para detectar y corregir el error. La frecuencia de ocurrencia de los errores de secuenciación es muy baja, alrededor del 1% en las lecturas de la mayoría de las tecnologías NGS (5).
Repeticiones: Normalmente los genomas contienen gran cantidad de regiones repetidas, las cuales crean problemas de alineación fundamentalmente por las limitadas longitudes de las lecturas generadas por las máquinas NGS, en esencia entre más cortas sean tales lecturas, mayor es la probabilidad de que se concuerden erróneamente en localidades del genoma que se repiten. Este punto se trata en varias formas, dependiendo del programa usado, muchos programas utilizan la primer concordancia para la alineación, mientras que otros descartan áreas repetitivas completamente, lamentablemente ninguno de los métodos resuelve el problema, recurriendo a soluciones más sofisticadas como el uso de lecturas por pares.
PROGRAMAS DE ALINEACIÓN
El desarrollo acelerado de las tecnologías NGS ha provocado, en los últimos años, el surgimiento de gran cantidad de software para la alineación de lecturas cortas. Al respecto existen diferentes trabajos que han revisado y comparado desde diversos puntos de vista tales aplicaciones, el más completo es el de Fonseca y equipo (14) quienes presentan un análisis comparativo de 60 programas de este tipo. Otros trabajos como los de las referencias (15), (16) y (17), limitan el estudio a un pequeño subconjunto de tales programas (los más utilizados y referenciados al momento de escribir cada artículo), pero analizándolos en forma más profunda. En realidad, es difícil poder aseverar que un programa sea mejor que otro, cada uno muestra características particulares que lo hace apropiado a determinada plataforma de secuenciación (illumina, Roche 454, etc.), o a un tipo de dato específico (ADN, bisulfito, miARN, ARN, etc.). También difieren en la forma en que reportan los resultados de la alineación, el número de desapareos, inserciones y supresiones (indels) permitidas, la habilidad de permitir indels consecutivos, la capacidad de alinear lecturas apareadas (PE), el uso de información de calidad (QA), entre otras variantes. La Tabla 3 resume las características de los programas de alineación más representativos en la actualidad.
Por otra parte, algorítmicamente la mayoría de los programas de alineación de lecturas cortas utilizan solo dos estrategias: Tablas Hash y Transformada de Burrows-Wheeler (TBW), esta última siendo el resultado de la evolución de los árboles y arreglos de sufijos. Ambas estrategias serán discutidas en los apartados siguientes.
]]> Algoritmos basados en Tablas Hash
Para lograr eficiencia, todos los métodos deben basarse en un tipo de pre-computo. Tablas Hash utiliza la idea de compilar una lista de todas las palabras de longitud l y determinar una sola vez sus posiciones en el genoma de referencia. Posteriormente puede utilizarse un algoritmo de hasheo, para transformar una lectura corta en una clave que permita una búsqueda rápida. Aunque esta idea es teóricamente concebible, falla en la práctica por el uso excesivo de memoria en la computadora, además de que, el esquema básico aún no considera alineaciones inexactas. Una posible solución a este problema es el uso de k - mers (subcadenas de longitud k, con k < l), eligiendo un valor de k mucho menor a l se pueden almacenar todos los k- mers traslapados, que aparezcan en la referencia en una lista, tal como se observa en la parte a de la Figura 5. Posteriormente cada lectura puede dividirse en semillas de longitud k y buscarse sobre la lista (ver Figura 5, parte b).
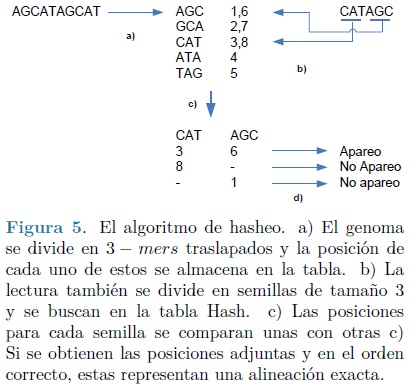
Si las semillas de una lectura se encuentran en la lista, en el orden correcto y adjuntas una a otra, la lectura existe en el genoma (parte c y d de la Figura 5). En términos del espacio utilizado, ahora el problema es más tratable ya que hay a lo mucho 4k diferentes k- mers en el genoma. Aún así este algoritmo no permite alineación inexacta.
El algoritmo previo puede modificarse para alinear lecturas permitiendo errores (desapareos e indels). Suponga que se permiten dos errores durante la alineación, en tal caso se puede asegurar que a lo mucho dos de los k- mers en que se ha dividido la lectura contendrán errores y el resto de estos no los contendrán, alineándose en forma exacta al genoma (ver Figura 6), lo anterior se conoce como el principio de las cajas o del palomar. Los k - mers que se alinean perfectamente al genoma constituyen una "semilla", y al aplicar un algoritmo más preciso como los basados en programación dinámica en la vecindad de esta semilla, es posible alinear la lectura conteniendo errores. Esta estrategia de dos pasos llamada "siembra y extiende" se implementa en muchas herramientas tales como MAQ (18), PASS (19), SSAHA2 (20), SOAP (21), RMAP (22) y SeqMap (23).

El problema fundamental del algoritmo siembra y extiende es que las lecturas necesitan dividirse en subcadenas cada vez más pequeñas cuando el número de errores permitidos se incrementa. Estas subcadenas tan pequeñas suelen producir errores en la fase de sembrado, puesto que tienen mayor probabilidad de alinearse equivocadamente en múltiples regiones del genoma (falsos positivos), es por esta razón que no es común el uso de semillas de menos de 10 nucleótidos. Para sobrellevar el problema, algunos programas como ZOOM (24), GASSST (25), BFAST (26) y SHRiMP2 (27) han recurrido al uso de semillas espaciadas, es decir semillas conteniendo posiciones "no importa", en las cuales el algoritmo no checa el tipo de nucleótido presente. Por ejemplo, indicando a x como la posición no importa, la semilla AGTCxGA es capaz de alinearse a AGTCAGA, AGTCCGA, etc. Es evidente que utilizar un conjunto de semillas espaciadas en lugar de una semilla regular incrementa la sensibilidad del método, aunque tiene el efecto lateral de incrementar el tiempo de cómputo.
Las herramientas basadas en el algoritmo siembra y extiende gastan la mayor parte del tiempo en la etapa de extensión, implementada usualmente mediante algún algoritmo de programación dinámica, como el Smith-Waterman (28) o el Needleman-Wush (29), los cuales permiten en forma natural alineaciones locales con indels y desapareos. Para superar la complejidad temporal de esta etapa, los programas utilizan optimizaciones comunes de los algoritmos de programación dinámica y en algunos casos, como en SHRiMP2, comandos especiales del CPU para paralelizar el trabajo. Otros métodos agregan un paso intermedio entre la siembra y la extensión, tal es el caso de GASSST, el cual cada vez que encuentra una semilla, compara rápidamente la región vecina con el resto de la lectura, usando un algoritmo mucho más rápido que los de programación dinámica. La etapa agregada denominada filtro, consiste en calcular la distancia de Euler, la cual halla el número de letras de cada tipo en las regiones comparadas, si por ejemplo se trata de alinear una región conteniendo tres As, con una región que contiene cinco As, es evidente la existencia de al menos dos errores en la alineación, de esta forma las regiones que no cumplan con este filtro pueden ser descartadas disminuyendo notablemente la carga de la etapa de alineación. Esta variante del algoritmo se llama apropiadamente siembra, filtra y extiende.
]]>Algoritmos basados en TBW
La transformada de Burrows-Wheeler (TBW), presentada originalmente en (30), es un algoritmo que transforma una cadena de caracteres en otra mucho más fácil de comprimir. El re-ordenamiento se hace en tal modo que la cadena resultante agrupa los caracteres similares en la cadena original, de esta forma puede comprimirse fácilmente usando codificación move-to-front y codificación run-length. No obstante, recientemente la TBW encontró una aplicación diferente a la compresión de datos, luego de que Ferragina y Manzini (31), mostraron como la TBW hereda todas las propiedades y potencia comput acional de los arreglo s de sufijos, pero requiriendo mucho menos espacio en memoria que estos. De esta forma numerosos programas de alineación recientes como SOAP2 (32), Bowtie (33) y BWA (34), utilizan la TBW como estructura de datos principal.
Transformada de Burrows-Wheeler
Más que establecer una definición rigurosa de la TBW, en lo que sigue se presenta el algoritmo mediante el ejemplo simple mostrado en la Figura 7. Sea R = TAGACAGA la cadena de entrada a transformar, Σ = {A, C, G, T}, el alfabeto sobre el cual es construida la cadena, y $ un carácter especial lexicográficamente menor que todos los otros caracteres en Σ. El algoritmo comienza agregando el carácter especial $ a la cadena de entrada R4, de esta forma obteniendo la secuencia R' = TAGACAGA$, el carácter especial es necesario para poder revertir la transformación y mantener la compatibilidad con el arreglo de sufijos equivalente. Posteriormente se generan todas las rotaciones de R' y se colocan en un arreglo conceptual como se muestra en la Figura 7a, Observe como en tal arreglo pueden identificarse todos los sufijos posibles de la cadena R. Finalmente las rotaciones se ordenan lexicográficamente como se muestra en la Figura 7b. La última columna del arreglo es la transformada buscada, en el ejemplo: L(i) = AGGCTAAA$. En los arreglos de la Figura 7 las variables i y S(i) representan el índice de los arreglos, y el arreglo de sufijos correspondientes, ambas colocadas solamente por claridad.

A pesar de que el uso asintótico de memoria es 0(|R|), la cantidad de almacenamiento que se requiere para guardar la TBW es significantemente más pequeño que el necesario para un arreglo de sufijos. Almacenar la TBW requiere el mismo espacio que el texto original, ya que esta es solo una permutación de dicho texto. Adicionalmente, en el caso tratado, el alfabeto consiste de solo cuatro letras, de tal forma que cada una de estas puede representarse mediante dos bits, así para almacenar la TBW del genoma humano se requiere ~ 2 * 3 * 109 bits, en lugar de los ~ 32 * 3 * 109 bits del arreglo de sufijos del mismo, suponiendo que cada entero utiliza 32 bits, como en la mayoría de las aplicaciones
]]> Los índices de FM
En el 2000, Paolo Ferragina y Geovanni Manzini publicaron un artículo (31) en el que se describía como la TBW junto con algunas estructuras de datos auxiliares, podrían usarse como un índice de R eficiente en espacio, a lo que llamaron índices de FM. La clave es el principio denominado Último-Primero enunciado a continuación:
Lema: (Propiedad de mapeo Último-Primero) La j-ésima ocurrencia de una letra en particular X en la última columna (L) de la matriz TBW, es la misma letra como la j-ésima ocurrencia de X en la primera columna (F).
Suponga que se desea buscar las ocurrencias del patrón P = AGA en la cadena de referencia R del ejemplo anterior Puesto que al igual que el arreglo de sufijos, la matriz TBW está ordenada lexicográficamente, las filas conteniendo a P como un prefijo se encontrarán de manera consecutiva El procedimiento de búsqueda se basa en el índice formado por F, L y un vector que contiene en cada posición i el número de veces que el carácter L[i] ha aparecido desde el inicio hasta esa fila e incluida esta, denominado vector de rango (Figura 8) La búsqueda inicia, encontrando las filas que comienzan con el sufijo más corto de P, A en este caso, lo anterior es trivial puesto que la primera columna es parte del índice, estas filas son aquellas en el intervalo [1,4] (Figura 8a). A continuación puede agregarse el siguiente carácter a la búsqueda formando el segundo sufijo más corto del patrón: GA. El espacio de búsqueda ahora está limitado al intervalo [1,4] hallado previamente. Observando la columna L en ese rango puede notarse que existen dos filas en las cuales G precede a A (filas 1 y 2), y de acuerdo al vector de rango, estas corresponden a la primera y segunda aparición de G en L. Finalmente, por el mapeo LF puede determinarse que esos mismos caracteres se localizan en las filas 6 y 7 de F (primera y segunda aparición de G en F), en otras palabras, se ha obtenido un nuevo intervalo de búsqueda: [6,7], como se muestra en la Figura 8b. Posteriormente se agrega el último carácter del patrón buscado, formando el sufijo AGA. Nuevamente revisando L en el intervalo encontrado, puede observarse que existen dos filas en las cuales A precede al prefijo GA (filas 6 y 7), y al aplicar el mapeo LF se halla que esos mismos caracteres en L se encuentran en las posiciones 3 y 4 respectivamente, el cual efectivamente es el intervalo en el que aparece el prefijo buscado (Figura 8c).
Observe como en la Figura 8, se incluyó la matriz TBW completa solo por claridad, interviniendo en este ejemplo inicial solo la primera y la última columna de la misma. Este procedimiento es llamado búsqueda hacia atrás. En resumen, consiste en aplicar el mapeo LF repetidamente para encontrar el rango de filas prefijadas por sufijos de P alargados progresivamente, hasta que el rango llegue a estar vacío, lo cual ocurre si P no existe en R, o hasta que se termine con los sufijos, en cuyo caso el tamaño del rango es el número de veces que P ocurre en R.
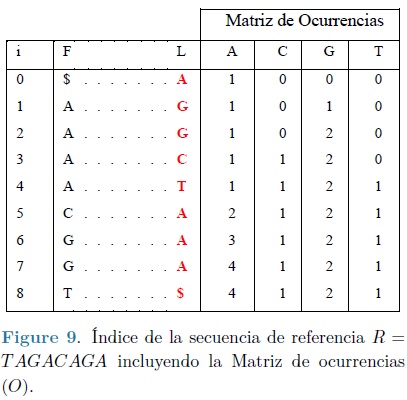
La complejidad temporal del mismo puede mejorarse si en lugar de almacenar un vector de rangos se almacena la matriz de ocurrencia O, en cada fila de tal matriz se guarda un entero para cada carácter del alfabeto igual al número de veces que el carácter ha aparecido en L, hasta ese número de fila e incluida esta, lo anterior puede observarse en la Figura 9. Ahora en lugar de escanear la última columna, simplemente se busca el carácter apropiado en los extremos izquierdo y derecho del rango actual, si no hay diferencias entre las dos búsquedas, el carácter no ocurre. Si hay una o más ocurrencias del carácter, la búsqueda dará los rangos de esas ocurrencias.
El algoritmo de búsqueda formalizado, adaptado de la referencia (31), se presenta en la Figura 10. En este interviene la matriz de ocurrencias O previamente definida y el arreglo unidimensional C de longitud |Σ| en donde C['x'] representa el número de caracteres en R que son lexicográficamente más pequeños que 'x' sin considerar el carácter especial $. En el ejemplo particular que se ha seguido este corresponde a:


Las variables k y l representan los extremos actuales del intervalo inferior y superior respectivamente, P el patrón buscado, i un apuntador y σ una variable que contiene el carácter del patrón procesado actualmente. Las líneas 2 a 4 inicializan el proceso colocando el índice i apuntando al carácter más a la derecha del patrón y los indices k y l indican el intervalo más grande posible de búsqueda (0 y |R|-1, respectivamente).
Las líneas 5 a la 10 forman el núcleo del proceso, básicamente el bucle toma uno a uno los carácteres del patrón de derecha a izquierda, aplicando el mapeo último a primero en las líneas 7 y 8 para definir el nuevo intervalo de búsqueda, tal como se explicó previamente. Observe como el término C[σ] + 1, en realidad señala la primer posición de aparición del carácter a en la primera fila y es adicionada al temrino O [σ, k - 1], que representa el número de ocurrencias del mismo carácter hasta la fila k -1, para obtener el mepeo deseado. Finalmente el algoritmo retorna el intervalo [k,j] si k ≤ l o un intervalo vacío en caso contrario (líneas 10 a 15).
La complejidad temporal del algoritmo es O(|P|), el hecho de que la complejidad en tiempo no dependa del tamaño de la cadena de referencia es sumamente ventajoso para este tipo de aplicaciones, en donde secuencias cortas se alinean a cadenas extremadamente largas tales como el genoma humano.
Por otra parte, el esquema que acaba de presentarse no toma en cuenta búsquedas aproximadas. No obstante es posible adaptar el algoritmo para conseguirlo, habitualmente esto se logra mediante uno de los siguientes métodos:
• Expresando la búsqueda aproximada en términos de muchas búsquedas exactas. La idea básica es crear la vecindad de palabras del patrón P, que se encuentren a una distancia de edición k (siendo k el número de diferencias permitidas en la búsqueda). Una vez generada la vecindad simplemente se busca cada palabra sobre el índice y se guarda cada acierto como una concordancia aproximada en esa ubicación dentro del texto. Lo anterior puede sonar muy simple sin embargo, el tamaño de la vecindad crece abruptamente al incrementar el número de diferencias permitidas. El tamaño de la vecindad de una palabra está limitado a O(|P|k|Σ|k), así si se intenta crear la vecindad de una lectura corta de ADN con 32 bases nucleicas de longitud y permitiendo 3 diferencias, el número de palabras sería de 2,985,984. Debido a la explosión combinatoria, el método solo es práctico para valores pequeños de k.
• Adoptando la estrategia siembra y extiende. La técnica es idéntica a la presentada en la sección dedicada a tablas hash. De acuerdo a los parámetros de la búsqueda algunas sub-cadenas del patrón se localizan en forma exacta en el índice y se suman a una lista de concordancias candidatas, las cuales son posteriormente examinadas alrededor de sus posiciones mediante un algoritmo más preciso, validando o no la concordancia del patrón completo.
El lector interesado puede encontrar en (35), una descripción más detallada acerca de posibles modos de hallar concordancias aproximadas. Debe notarse que, distinto a lo que pasa con tablas Hash, en tal esquema de búsqueda no se modifica el índice al permitir diferencias, la extensión es siempre realizada algorítmicamente, pero desarrollando una búsqueda más compleja.
]]>ACELERACIÓN HARDWARE
Los datos NGS consisten de miles de millones de lecturas cortas, por lo que la mayoría de los programas de alineación, tienen soporte para ejecución paralela en sistemas con memoria distribuida (clústeres compuestos por múltiples computadoras) y/o usando memoria compartida (equipos de cómputo con múltiples núcleos). Aún así, los tiempos para terminar la tarea, siguen siendo muy elevados (14) (36). Por ejemplo, en (14), los autores comparan empíricamente la velocidad computacional y requisitos de memoria de algunos alineadores representativos, utilizando como referencia el genoma humano (Homo Sapiens, assembly GRCh37). Al alinear un millón de lecturas cortas de 100 nucleótidos de longitud a tal genoma, los resultados muestran que los alineadores BWA, Bowtie y SOAP2 (más eficientes en la comparación), utilizan en promedio 97, 169 y 176 minutos, mientras requieren 7.6, 5 y 5.3 Gb de memoria RAM respectivamente. En contraste la producción de esta misma cantidad de lecturas mediante un secuenciador NGS, por ejemplo Hiseq, es de aproximadamente 5 minutos (Ver tabla 2). Esta diferencia entre tiempos de generación y alineación, ha creado la necesidad de nuevas formas de acelerar los programas de alineación, abriendo una nueva área de aplicación a la aceleración hardware.
En cómputo, la aceleración hardware es el uso de circuitos especializados para desarrollar alguna función más rápida que la lograda mediante la ejecución del algoritmo sobre un procesador de propósito general. En la actualidad las alternativas más generalizadas para implementarla son: los circuitos integrados de aplicación específica (ASIC), las unidades de aceleración gráfica (GPUs), y los dispositivos lógicos programables en el campo (FPGAs), todas estas pueden permitir que miles de unidades trabajen simultáneamente logrando el procesamiento paralelo masivo de los datos. Esta técnica aunque pueda parecer universalmente efectiva, solo toma ventaja cuando las funciones incluyen cálculos repetitivos e independientes, tal es el caso del proceso de alineación de ADN en donde millones de lecturas se alinean al genoma de referencia de forma independiente unas de otras.
Los ASIC tienen un desempeño superior al de sus contrincantes, permitiendo el mínimo consumo de potencia y la más alta velocidad de procesamiento, sin embargo a diferencia de los GPUs y los FPGAs no pueden reprogramarse, este factor limitante resulta en un incremento en el tiempo de desarrollo y en los costos de ingeniería no recurrente (NRE). Por su parte, las GPUs son coprocesadores paralelos muy económicos con una enorme penetración en el mercado, dedicados al procesamiento de gráficos u operaciones de coma flotante, para aligerar la carga de trabajo del procesador central en aplicaciones como los videojuegos o aplicaciones 3D interactivas, tienen una memoria con gran ancho de banda y un número elevado de núcleos programables, con miles de hilos hardware ejecutando programas en un modo Programa Único Múltiples Datos (SPMD), son flexibles y fáciles de programar usando lenguajes de alto nivel y APIs las cuales abstraen la mayoría de los detalles del hardware. Comparado a la arquitectura hardware fija del GPU, los FPGAs son esencialmente arreglos densos de bloques lógicos programables, prefabricados y que el desarrollador puede (re)configurar módulo a módulo para darle la funcionalidad deseada. De esta forma se tiene el control de los compromisos entre recursos, desarrollo y nivel de paralelismo, obteniendo diseños superiores en desempeño a los basados en GPUs y cercanos en eficiencia a un ASIC (37). Lamentablemente aunque algunos vendedores proveen núcleos con propiedad intelectual (IP) que ofrecen la mayoría de las funciones de procesamiento más comunes, la configuración de un FPGA requiere el uso de lenguajes de descripción de hardware los cuales son más complejos de utilizar que los lenguajes comunes de alto nivel.
Existen en la literatura varios trabajos que se han enfocado en acelerar los algoritmos de alineación de lecturas mediante hardware especializado. SOAP3 presentado en (38) es una presentación GPU habilitada por CUDA del popular software de alineación SOAP. Sus creadores reportan aceleraciones de 7.5X comparado a BWA, no obstante, su diseño no soporta alineación con espacios. CUSHAW presentado en (39) es también una implementación basada en CUDA de los índices de FM, no permite alineaciones con espacios y reporta una aceleración de 6X comparado a la versión multihilos de BWA. En (40) los autores presentan SHEPARD una implementación FPGA de un algoritmo basado en tablas Hash sobre la plataforma de alto desarrollo Convey HC1. Reportan velocidades extremadamente altas comparadas con herramientas software como BWA, Bowtie y MAQ. No obstante, su arquitectura soporta únicamente alineación exacta y es capaz de alinear solo un bajo porcentaje de las lecturas entrantes, además de utilizar una elevada cantidad de memoria. En (41) se presenta la primer implementación de alineación exacta mediante los índices de FM. Al comparar sus resultados con el software de alineación BOWTIE, obtienen aceleraciones mayores a 100X, sin embargo al almacenar la tabla de ocurrencia y el vector de frecuencia en módulos BRAM, su aplicación se limita a cadenas de ADN relativamente cortas. Una mejora importante al trabajo de Fernández se reporta en (42), su método consiste de dos componentes claves, un alineador de lecturas exacto para el grueso del proceso de alineación y un alineador aproximado para los casos restantes. La arquitectura se implementa mediante configuración dinámica en el FPGA, maximizando el uso de recursos del mismo. Muestran que una implementación particular de su método utilizando un solo FPGA puede ser 293 veces más rápido que BWA sobre un procesador Intel X5650, y 134 veces más rápido que SOAP3 en una tarjeta NVIDIA GTX 580 GPU. En (43), se presenta un acelerador hardware del software BWA, los autores reportan una aceleración de 10X. El diseño únicamente está probado en una referencia de pocos miles de nucleótidos, resultando en una velocidad de procesamiento menor que la utilizada usando referencias más grandes. A excepción de SOAP3 y CUSHAW, el resto de los diseños son versiones preliminares y los resultados comparativos con alineadores software son estimaciones a partir de la razón de procesamiento.
DISCUSIÓN Y CONCLUSIONES
La secuenciación de ADN es un área activa de investigación a nivel mundial. Por una parte, los fabricantes de máquinas NGS se han enfocado en el uso altamente masivo de dispositivos de fluidos ópticos y electrónicos, todos a escala microscópica, logrando la producción de enormes cantidades de lecturas por corrida, no obstante están limitadas a secuenciar solo fragmentos cortos de ADN, por lo que el reto es incrementar la longitud de tales fragmentos a miles o millones de nucleótidos, y en un futuro no muy lejano de genomas completos sin necesidad de segmentar el mismo. Por otra parte los desarrolladores de software buscan continuamente algoritmos más eficientes en tiempo y espacio para llevar a cabo el ensamble de fragmentos, utilizando instrucciones especiales para ejecución paralela en múltiples núcleos y/o procesadores. Un tercer grupo de investigadores incursiona en acelerar los algoritmos de ensamble mediante alternativas diferentes a la ejecución sobre procesadores de propósito general, en este sentido existen dos opciones fundamentales, las GPUs y los FPGAs, ambas ofrecen miles de núcleos hardware trabajando en paralelo. Los primeros son una opción muy económica y son relativamente fáciles de programar mientras que los segundos ofrecen una mayor efectividad y flexibilidad a costa de mayor complejidad en su configuración y precio elevado. A pesar de estos esfuerzos existe aún la diferencia entre el tiempo de producción y de ensamble de fragmentos en un orden superior a 10x, convirtiéndose esta última etapa en el cuello de botella del proceso de análisis de ADN.
]]> RECONOCIMIENTOS
Este proyecto fue apoyado por el programa para el mejoramiento del profesorado (PROMEP) y la Universidad del Istmo (UNISTMO) a través de la beca con folio UNISTMO-003.
REFERENCIAS
1. Frese, K.S., Katus, H.A. and Meder, B. "Next-Generation Sequencing: From understanding biology to personalized medicine". Biology, Vol. 2, pp. 378-398, 2013. [ Links ]
2. Sanger, F., Nicklen, S. and Coulson, A.R. "DNA sequencing with chain-terminating inhibitors". PNAS, Vol. 74, No. 12, pp. 5463-5467, 1977. [ Links ]
3. Maxam, A. and Gilbert, A. "A new method for sequencing DNA". PNAS, Vol. 74, No. 2, pp. 560-564, 1977. [ Links ]
]]>4. Venter, C., et al. "The sequence of the human genome". Science, Vol. 291, pp. 1304-1351, 2001. [ Links ]
5. Liu, L., et al. "Comparison of Next generation sequencing systems". Journal of Biomedicine and Biotechnology, pp. 1-11, 2012. [ Links ]
6. Myllykangas, S., Buenrostro, J. and Ji, H.P. "Overview of sequencing technology platforms". [book auth.] Naiara Rodríguez Ezpeleta, Michael Hackenberg and Ana M. Aransay. Bioinformatics for high throughput sequencing. s.l. : Springer, 2012, pp. 11-25. [ Links ]
7. Quail, M.A., et al. "A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers". BMC Genomics, Vol. 13. No. 341, 2012. [ Links ]
8. Pop, M. "Shotgun sequence assembly". Advances in Computers, Vol. 60, pp. 193-248, 2004. [ Links ]
]]>9. Kim, R.Y., et al. "The future of personalized care in colorectal cancer". Personalized Medicine, Vol. 8, No. 3, pp. 331-345, 2011. [ Links ]
10. Li, Z., et al. "Comparison of the two major classes of assembly algorithms". Briefings in Functional Genomics, Vol. 11, No. 1, pp. 25-37, 2012. [ Links ]
11. Miller, J.R., Koren, S. and Sutton, G. "Assembly algorithms for next-generation sequencing data". Genomics, vol. 95, no. 6, pp. 315-327, 2010. [ Links ]
12. Altschul, S et al. "Basic local alignment search tool". Journal of Molecular Biology, Vol. 215, No. 3, pp. 403-410, 1990. [ Links ]
13. Muse, S. Genomics and bioinformatics. [book auth.] John D. Enderle, Susan M. Blanchard and Joseph D. Bronzino. Introduction to Biomedical Engineering. 2. s.l. : Elsevier, 2005, pp. 799-831. [ Links ]
]]>14. Fonseca, N.A, et al. "Tools for mapping high-throughput sequencing data". Bioinformatics, Vol. 28, No. 24, pp. 3169-3177, 2012. [ Links ]
15. Shang, J., et al. "Evaluation and comparison of multiple aligners for next-generation sequencing data analysis". BioMed Research International, Vol. 2014. [ Links ]
16. Ruffalo, M., LaFramboise, T. and Koyutürk, M. "Comparative analysis of algorithms for next-generation sequencing read alignment". Bioinformatics, Vol. 27, No. 20, pp. 2790-2796, 2011. [ Links ]
17. Li, H. and Homer, N. "A survey of sequence alignment algorithms for next-generation sequencing". Briefings in Biointormatics, Vol. 2, No. 5, pp. 473-483, 2010. [ Links ]
18. Li, H., Ruan, J. and Durbin, R. "Mapping short DNA sequencing reads and calling variants using mapping quality scores". Genome Research, Vol. 18, pp. 1851-1858, 2008. [ Links ]
]]>19. Campagna, D., et al. "PASS: a program to align short sequences". Bioinformatics, Vol. 25, No. 7, pp. 967-968, 2009. [ Links ]
20. Ning, Z., Cox, A. and Mullikin, J. "SSAHA: A fast search method for large DNA databases". Genome Research, Vol. 11, No. 10, pp. 1725-1729, 2001. [ Links ]
21. Li, R., et al. "SOAP: short oligonucleotide alignment program". Bioinformatics, Vol. 24, No. 5, pp. 713-714, 2008. [ Links ]
22. Smith, A.D., Xuan, Z. and Zhang, M.Q. "Using quality scores and longer reads improves accuracy af Solexa read mapping". BMC Bioinformatics, Vol. 9, No. 128, 2008. [ Links ]
23. Jiang, H. and Wong, W. "SeqMap: mapping massive amount of oligonucleotides to the genome". Bioinformatics, Vol. 24, No. 20, p. 2395, 2008. [ Links ]
]]>24. Lin, H., et al. "Zoom! Zillions of oligos mapped". Bioinformatics, Vol. 24, No. 21, pp. 2431-2437, 2008. [ Links ]
25. Rizk, G. and Lavenier, D. "GASSST: Global alignment short sequence search tool". Bioinformatics, Vol. 26, No. 20, pp. 2534-2540, 2010. [ Links ]
26. Homer, N., Merriman, B. and Nelson, S. "BFAST: an alignment tool for large scale genome resequencing". PLoS ONE, Vol. 4, 2009. [ Links ]
27. David, M., et al. "SHRiMP2", Bioinformatics, Vol. 27, No. 7, pp. 1011-1012, 2011. [ Links ]
28. Smith, T.F. and Waterman, M. S. "Identification of common molecular subsequences". Journal of Molecular Biology, Vol. 147, No. 1, pp. 195-197, 1981. [ Links ]
]]>29. Needleman, S.B. and Wunsch, C.D. "A general method applicable to the search for similarities in the aminoacid sequence of two proteins". Journal of Molecular Biology, Vol. 48, No. 3, pp. 443-453, 1970. [ Links ]
30. Burrows, M. and Wheeler, D.J. "A block sorting lossless data compression algorithm". Systems Research Center, Digital Equipment Corporation. Palo Alto, California : s.n., 1994. Reporte Técnico. 124. [ Links ]
31. Ferragina, P. and Manzini, G. "Opportunistic data structures with applications". Redondo Beach, CA: IEEE, 2000. Foundations of computer science. pp. 390-398. [ Links ]
32. Li, R., et al., et al. "SOAP2: an improved ultrafast tool for short read alignment". Bioinformatics, Vol. 25, No. 15, pp. 1966-1967, 2009. [ Links ]
33. Langmead, B., et al. "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome". Genome Biology, Vol. 10, No. 3, 2009. [ Links ]
]]>34. Li, H. and Durbin, R. "Fast and accurate short read alignment with Burrows-Wheeler transform". Bioinformatics, Vol. 25, No. 14, pp. 1754-1760, 2009. [ Links ]
35. Navarro, G., et al. "Indexing methods for aproximate string matching". IEEE Data Engineering Bulleting, Vol. 24, No. 4, pp. 19-27, 2001. [ Links ]
36. Schbath, S., et al. "Mapping reads on a genomic sequence: an practical comparative analysis". Statistics for systems biology group. Paris, Francia : s.n., 2011. Reporte técnico. 34. [ Links ]
37. Che, S., et al. "Accelerating compute-intensive applications with GPUs and FPGAs". Anahem, CA : IEEE, 2008. Application specific processors, SASP2008. pp. 101-107. [ Links ]
38. Liu, C.M., et al. "SOAP3: Ultra-fast GPU-based parallel alignment tool for short reads". Bioinformatics Advance Access Published, Vol. 28, No. 6, pp. 878-879, 2012. [ Links ]
]]>39. Liu, Y., Schmidt, B. and Maskell, D.L. "Cushaw: a cuda compatible short read aligner to large genomes based on the burrows-wheeler transform". BMC Research Notes, Vol. 5, No. 1, p. 27, 2012. [ Links ]
40. Nelson, C., et al. "Shepard: A fast exact match short read aligner". Formal integral 277 Methods and Models for Codesign (MEMOCODE), 2012 10th IEEE/ACM International Conference on. pp. 91-94. [ Links ]
41. Fernandez, E., Najjar, W. and Lonardi, S. "String matching in hardware using the FM-Index". Salt, Lake City, UT: IEEE, 2011. IEEE International Symposium on Field-Programmable Custom Computing Machines. pp. 218-225. [ Links ]
42. Arram, J., et al. "Reconfigurable acceleration of short read mapping". Seattle, WA : IEEE, 2013. 21st Annual International IEEE Symposium on Field-Programmable Custom Computing Machines. pp. 210-217. [ Links ]
43. Waidyasooriya, H.M., Hariyama, M. and Kameyama, M. "Implementation of a custom harwdware-accelerator for short-read mapping using Burrows-Wheeler Alignment". Osaka, Japan: IEEE, 2013. 35th Annual International Conference of the IEEE EMBS. pp. 651-654. [ Links ]
]]>