
Nuevas perspectivas en el síndrome de QT largo
New perspectives in long QT syndrome
Argelia Medeiros–Domingo,* Pedro Iturralde–Torres,** Samuel Cañizales–Quinteros,* Arturo Hernández–Cruz,*** Ma. Teresa Tusié–Luna*
* Unidad de Biología Molecular y Medicina Genómica, Instituto de Investigaciones Biomédicas. Universidad Nacional Autónoma de México e Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
** Instituto Nacional de Cardiología Ignacio Chávez.
]]> *** Instituto de Fisiología Celular, Universidad Nacional Autónoma de México.
Reimpresos:
Dra. Argelia Medeiros–Domingo
Unidad de Biología Molecular Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán
Vasco de Quiroga No. 15, Tlalpan
14000, México, D.F.
Tel. y fax 5655–0011.
Correo electrónico: argeliamed@yahoo.com
]]> Recibido el 17 de abril de 2006.
ABSTRACT
Long QT Syndrome (LQTS) is a cardiac channelopathy characterized by prolonged ventricular repolarization and increased risk to sudden death secondary to ventricular dysrrhythmias. Was the first cardiac channelopathy described and is probably the best understood. After a decade of the sentinel identification of ion channel mutation in LQTS, genotype–phenotype correlations have been developed along with important improvement in risk stratification and genetic guided–treatment. Genetic screening has shown that LQTS is more frequent than expected and interestingly, ethnic specific polymorphism conferring increased susceptibility to drug induced QT prolongation and torsades de pointes have been identified. A better understanding of ventricular arrhythmias as an adverse effect of ion channel binding drugs, allow the development of more safety formulas and better control of this public health problem. Progress in understanding the molecular basis of LQTS has been remarkable; eight different genes have been identified, however still 25% of patients remain genotype–negative. This article is an overview of the main LQTS knowledge developed during the last years.
Key words: Sudden death. Long QT Syndrome. Cardiac channelopathies. Arrhythmias.
RESUMEN
El síndrome de QT largo (SQTL) es una canalopatía que genera grave alteración en la repolarización ventricular predispone a arritmias malignas y muerte súbita. Fue la primera canalopatía arritmogénica descrita y quizá la mejor entendida hasta ahora. Transcurrida ya más de una década de la identificación de la primera mutación asociada al SQTL, se ha hecho evidente que este trastorno es mucho más frecuente de lo que inicialmente se pensaba; los avances en el conocimiento de la fisiopatología molecular de esta enfermedad han permitido hacer una correlación genotipo–fenotipo, optimizando el tratamiento y permitiendo estratificar el riesgo en forma precisa. Se ha logrado entender con mayor detalle los efectos adversos de distintas drogas que interactúan con los canales iónicos, permitiendo así generar fármacos más seguros y, en su defecto, monitorizar de cerca aquellos que a pesar de tener este efecto adverso, es necesaria su administración. Los avances son importantes pero no todo está dicho, 25% de los casos no tienen mutaciones en los genes descritos hasta la fecha, por lo que el SQTL continúa siendo motivo de investigación. El presente artículo constituye un resumen de los principales conceptos desarrollados en los últimos diez años que han sido cruciales en el manejo de esta enfermedad.
Palabras clave. Muerte súbita. Síndrome de QT largo. Canalopatías. Arritmias cardiacas.
]]>INTRODUCCIÓN
El síndrome de QT largo (SQTL) se caracteriza por una grave alteración en la repolarización ventricular; se manifiesta en el electrocardiograma (ECG) por un alargamiento en el intervalo QT que predispone a arritmias ventriculares malignas –taquicardia helicoidal– y muerte súbita. El cuadro clínico es muy variable: el paciente puede cursar asintomático, presentar síncope recurrente o bien muerte súbita como primera manifestación de la enfermedad. Inicialmente se catalogó como un trastorno raro, y efectivamente, la presentación grave de la enfermedad es esporádica. No obstante, los estudios genéticos han podido estimar una incidencia de mutaciones de 1/5,000. Tan sólo en Estados Unidos ocurren aproximadamente de 3,000 a 4,000 defunciones por año atribuidas a este síndrome. Se desconoce la incidencia de esta enfermedad en la población mexicana.
La muerte súbita se define como la ocurrida de forma inesperada y rápida (< 1 hora) en personas sin datos previos de enfermedad grave hasta el inicio de los síntomas.1,2 La principal causa es la cardiopatía isquémica, pero en 5–10% de los afectados en población general3 y el 30% en población joven4 (< 35 años), el corazón no muestra alteraciones macroscópicas. En este grupo de individuos hay que descartar una canalopatía arritmogénica como probable causa de muerte.
En 1989 se describió la primera enfermedad asociada a un canal iónico, la fibrosis quística. Desde entonces la lista de enfermedades asociadas a disfunción de estas cruciales estructuras de la membrana celular se ha incrementado en forma sorprendente, generando así lo que hoy conocemos como "canalopatías".
Los canales iónicos son proteínas transmembranales macromoleculares, ensambladas en complejos heteromultiméricos que incluyen un poro y subunidades accesorias o modulatorias. Los canales son cruciales para el funcionamiento apropiado de todos los tipos celulares. En el miocardio, el transporte coordinado de iones por los distintos canales, es responsable de generar el potencial de acción que hace latir el corazón en forma ordenada por lo menos cien mil veces al día (Figura 1).5
El SQTL, descrito desde 1953,6 fue reconocido como canalopatía en 1995.7,8 A raíz de este hallazgo pionero, fueron identificadas diversas entidades clínicas que predisponen a arritmias y/o muerte súbita. Entre las canalopatías que condicionan trastornos del ritmo o conducción, podemos citar, al síndrome de Brugada, el síndrome de QT corto, la taquicardia ventricular catecolaminérgica, la displasia arritmogénica del ventrículo derecho, la enfermedad en la conducción intraventricular cardiaca,9 enfermedad congénita del nodo sinusal, el bloqueo AV congénito y la fibrilación auricular familiar. A excepción de la displasia arritmogénica del ventrículo derecho, ninguna de estas enfermedades suele cursar con malformaciones cardiacas o alteraciones estructurales macrocoscópicas; con un interrogatorio clínico y análisis electrocardiográfico apropiados puede sospecharse en ellas.
PERSPECTIVA HISTÓRICA
En 1953, Herrlin y Móller6 reportaron el caso de un niño con sordera congénita y episodios repetitivos de síncope; por primera vez describieron los hallazgos electrocardiográficos del SQTL: intervalo QT anormalmente prolongado, alteraciones en la onda T y bradicardia. En 1957 Jervell y Lange Nielsen10 publicaron sus hallazgos en una familia de padres no consanguíneos con seis hijos, cuatro de los cuales tenían sordera congénita y episodios sincópales, tres de ellos tuvieron muerte súbita. El ECG de los casos mostraba un intervalo QT inusualmente largo. Ambos padres cursaban asintomáticos, tenían un ECG normal y no presentaban problemas de audición. En 1964, Romano y Ward publicaron en forma independiente un síndrome cardíaco familiar caracterizado por síncope recurrente, antecedente familiar de muerte súbita, prolongación del intervalo QT sin sordera neuronal.11 Los estudios genéticos posteriores mostraron que el síndrome descrito por Jervell y Lange Nielsen, que se acompaña de sordera neuronal congénita, corresponde a mutaciones homocigotas, lo que da lugar a fenotipos muy graves con alto riesgo de muerte súbita. Por otro lado, el síndrome conocido como Romano–Ward corresponde a mutaciones heterocigotas: los pacientes no presentan trastornos en la audición y la gravedad de la enfermedad es muy variable.
]]> En 1995 se describieron los primeros genes asociados al SQTL,7,12 fue la primera canalopatía cardiaca descrita como tal y quizá la mejor estudiada hasta la fecha. Se han identificado ya 8 loci que explican 70–75% de los casos. En 25–30% de los pacientes, no se encuentran alteraciones en los genes descritos.13
PRINCIPALES CANALES ASOCIADOS AL SQTL
Para fines de esta revisión, nos referiremos principalmente a los canales dependientes de voltaje que transportan selectivamente los iones K+ y Na+, pues con mayor frecuencia se ven implicados en el SQTL.
Canales de potasio I Kr e IKs
El corazón posee diversos subtipos de canales selectivos al transporte del ion K+ cuya apertura es dependiente de voltaje (canales Kv). Típicamente, la salida de potasio es responsable de la repolarización celular y el fin del ciclo excitación–contracción.14
Los canales IKr e IKs, participan en la fase 3 del potencial de acción, son tetrámeros formados por la asociación de cuatro α–subunidades (Figura 2).
Canales de sodio
Se han descrito nueve isoformas de canal de sodio dependiente del voltaje en el humano (Navl.1–1.9),20 en el corazón predomina la isoforma Navl.5. En 1992, Gellens, et al., clonaron y caracterizaron el gen (SCN5A) que codifica la isoforma cardiaca.21 A diferencia de los canales de potasio descritos previamente, los canales de sodio son monómeros, formados por una estructura principal o subunidad α, de 260 kDa, que abarca cuatro dominios homólogos (Figura 3). Cada dominio tiene seis segmentos conectados entre sí por asas intra y extracelulares. El 4° segmento tiene residuos cargados positivamente y corresponde al sensor de voltaje. Entre los segmentos 5 y 6 se encuentra la región formadora del poro del canal. Esta región es altamente selectiva para los iones de Na+, y cataliza su transporte a través de la membrana. Diversas subunidades auxiliares, denominadas β, modulan la subunidad a.22,23 Hasta el momento se han descrito cuatro subtipos de subunidades P que han sido numeradas en forma progresiva según el orden de su descubrimiento.24–27
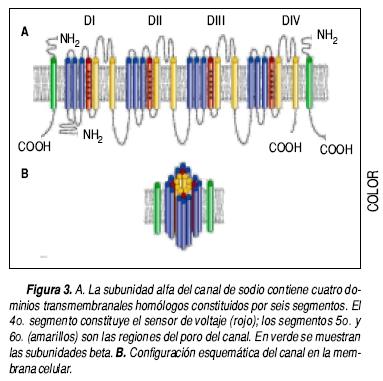
En el SQTL asociado a disfunción del canal de sodio Nav1.5, ocurre una inactivación inapropiada del canal, el cual permanece permeable a los iones Na+, cuando debería estar inactivado. Esta alteración ocasiona que durante la fase 2 exista una pequeña pero continua corriente despolarizante que prolonga la duración del potencial de acción.
CLASIFICACIÓN DEL SÍNDROME DE QT LARGO
La clasificación utilizada en la actualidad se fundamenta en el diagnóstico genético. Tenemos así los siguientes subtipos (Cuadro 1):
]]>Síndrome de QT largo tipo 1 (SQTL1)
Es el subtipo más frecuente (30–35% de los casos). El gen afectado es el KvLQT1(o KCNQ1) localizado en el cromosoma 11 (11p15.5), el cual codifica la subunidad a del IKs de canal de potasio. Estos pacientes suelen presentar episodios de taquicardia helicoidal al realizar ejercicio o al estimular el simpático (62%).28 La penetrancia en este subtipo es cercana a 60.4%, por lo que con frecuencia se observan portadores de la mutación con intervalo QTc dentro de límites normales. El potencial de acción se prolonga por una disminución de la corriente saliente de K+ durante fase 3.
Síndrome de QT largo tipo 2 (SQTL2)
El gen afectado es el KCNH2 o HERG localizado en el cromosoma 7 (7q35–36), el cual codifica la subunidad a del canal de potasio IKr (25–30% de los casos). En estos pacientes las arritmias se presentan con mayor frecuencia en respuesta a estrés emocional o estímulos auditivos súbitos –p.e. reloj despertador– (43%) y con menos frecuencia al ejercicio (13%).28 La penetrancia estimada es de 79.1%. Al igual que en el SQTL1, la disfunción de este canal, disminuye la corriente saliente de K+ durante la fase 3 del potencial de acción, prolongando así su duración.
Síndrome de QT largo tipo 3 (SQTL3)
El gen afectado es el SCN5A, que codifica para canal de sodio Nav1.5, localizado en el cromosoma 3 (3p21–24); es causante de la enfermedad en 5–10% de los casos. La inactivación defectuosa del canal, permite la entrada sostenida de Na+, durante la fase 2 del potencial de acción. Estos pacientes tienen un riesgo mayor de presentar arritmias malignas durante el reposo (sueño) o bradicardia.29 La penetrancia de las mutaciones en este gen es cercana a 90%.
Las mutaciones homocigotas en los subtipos SQTL1 y SQTL5 generan el síndrome de Jervell–Lange–Nielsen que cursa con sordera congénita y alto riesgo de muerte súbita. Afección a otros órganos.
]]>Síndrome de QT largo tipo 4 (SQTL4)
Es una variedad de SQTL extremadamente rara, el gen afectado es el ANKB, localizado en el cromosoma 4 (4q25–27), el cual codifica la síntesis de anquirina–β, perteneciente a una familia de proteínas estructurales de distribución ubicua, que vinculan proteínas membranales integrales con proteínas del citoesqueleto, entre otras, la bomba Na/K ATPasa, intercambiador Na/Ca y H/K ATPasa. Las mutaciones que causan pérdida de la función de anquirina–β, resultan en un incremento en la concentración de calcio intracelular, así como alteración en la expresión de N/K ATPasa e intercambiador Na/Ca; la conductancia a otros iones es afectada, favoreciendo no sólo la prolongación del intervalo QT, sino generando también despolarizaciones espontáneas. Se asocia con frecuencia a disfunción del nodo sinusal y bradicardia.30–32 Explica menos de 1% de los casos.
Síndrome de QT largo tipo 5 (SQTL5)
Es condicionado por cambios de secuencia del gen KCNE1 33 localizado en el cromosoma 21 (21q22.1–p22), codifica la síntesis de la subunidad β del canal IKs conocida también como subunidad minK que regula al canal IKa. Explica menos de 1% de los casos.
Síndrome de QT largo tipo 6 (SQTL6)
El gen afectado es el KCNE234 localizado en el cromosoma 21 (21q22.1). Codifica la subunidad β del canal de potasio, conocida también como subunidad MiRPl que regula al canal IKr. Explica menos de 1% de los casos.
]]> MUTACIONES QUE ASOCIAN SQTL Y ENFERMEDAD SISTÉMICA
Síndrome de QT largo tipo 7 (SQTL7)
Es causado por mutaciones en el gen KCNJ2 localizado en el cromosoma 17 (17q23) que codifica la síntesis del canal Kir 2.1; este canal participa en la fase 4 del potencial de acción. Su alteración da lugar al síndrome de Andersen, desorden autosómico dominante que se caracteriza por SQTL, parálisis periódica, desarrollo esquelético anormal, arritmias ventriculares del tipo de la extrasistolia ventricular frecuente con susceptibilidad particular a presentar fibrilación ventricular, sobre todo en el sexo femenino. Su expresividad es variable, lo que complica el diagnóstico oportuno de la enfermedad.35,36 Explica menos de 0.5% de los casos.
Síndrome de QT largo tipo 8 (SQTL8)
Descrito recientemente, resulta de mutaciones en el gen que codifica el canal de calcio tipo L Cav1.2. Ocasiona el síndrome de Timothy,37 caracterizado por malformaciones cardíacas, deficiencia inmunológica, hipoglucemia intermitente, trastornos cognitivos, incluso autismo; fusiones interdigitales y QT largo que predispone a arritmias cardíacas y muerte súbita.38 Explica menos de 0.5% de los casos.
MUTACIONES COMPUESTAS
Hasta en 8% de los casos con SQTL se pueden encontrar variantes en más de un gen. El intervalo QTc en estos casos suele ser muy prolongado, la incidencia de arritmias es alta con importante riesgo de muerte súbita.39
]]> Mutaciones de la variedad Jervell Lange–Nielsen
Corresponde a mutaciones homocigotas o heterocigotas compuestas en los genes KCNQ1 y/o KCNE1 que codifican la corriente IKs. En forma característica, se asocia a sordera congénita; los pacientes suelen tener un QTc > 500 ms, síncope recurrente por taquicardia helicoidal y alto riesgo de muerte súbita. En el tipo homocigoto, los padres de los pacientes con esta variedad son heterocigotos para la misma mutación y manifiestan una enfermedad menos grave o incluso pueden ser asintomáticos.40
De acuerdo con la clasificación anterior podemos concluir que los subtipos SQTL1, SQTL2 y SQTL3 son los más frecuentes y por lo tanto han sido mejor caracterizados clínicamente, con frecuencia nos referiremos a estas tres variantes exclusivamente.
Diagnóstico del SQTL
En 1993, Schwartz PJ, et al., publicaron los criterios diagnósticos para el síndrome de QT largo41 (Cuadro 2), los cuales son vigentes aún hoy en día.

Características electrocardiográficas Intervalo QT
]]> Un estudio multicéntrico reciente expone que al interpretar un trazo electrocardiográfico < 40% de los médicos no cardiólogos, < 50% de los cardiólogos y > 80% de los arritmiólogos, supieron medir correctamente este intervalo.42El intervalo QT se puede medir con mayor facilidad en las derivaciones II, VI, V3 o V5, pero se debe examinar el ECG de 12 derivaciones en busca del intervalo QT más largo (Figura 4).43 Convencionalmente, se utiliza la fórmula de Bazzet para corregir la duración del intervalo de acuerdo con la frecuencia cardiaca (QTc = 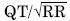 , expresado en segundos). Un intervalo QTc > 0.44 seg en los hombres y > 0.46 seg en las mujeres, debe considerarse anormal. No se ha llegado a un consenso respecto al límite normal inferior de este intervalo, y no había sido motivo de preocupación hasta la descripción reciente del síndrome de QT corto 44 que también se asocia a muerte súbita.
, expresado en segundos). Un intervalo QTc > 0.44 seg en los hombres y > 0.46 seg en las mujeres, debe considerarse anormal. No se ha llegado a un consenso respecto al límite normal inferior de este intervalo, y no había sido motivo de preocupación hasta la descripción reciente del síndrome de QT corto 44 que también se asocia a muerte súbita.
Dispersión del intervalo QT
La dispersión del intervalo QT refleja las diferencias regionales en la repolarización ventricular. La repolarización en el corazón es normalmente heterogénea debido a la diferencia en la duración del potencial de acción en las diferentes regiones (ventrículo derecho us. izquierdo o epicardio us. endocardio). Estas variaciones están dadas por la irregular distribución de diversos canales iónicos.45 La dispersión del intervalo QT es la variación del intervalo QT latido a latido, es más amplia cuando se comparan dos derivaciones que evalúan regiones diferentes y suele no variar significativamente en la misma derivación. La diferencia entre el QT máximo y mínimo se ha reportado en 48 ± 18 mseg en sujetos normales; en pacientes con SQTL esta dispersión se encuentra incrementada y la magnitud de este incremento se ha relacionado con un riesgo mayor de presentar arritmias ventriculares malignas;46 también es un parámetro de utilidad para valorar la eficacia terapéutica.47,48
Anormalidades morfológicas en la onda T
Los pacientes con SQTL pueden presentar múltiples alteraciones en la onda T 49: alternancia en la polaridad, apariencia bifásica, variaciones en la amplitud, muescas, entre otras. Estas anormalidades pueden permitir sospechar el subtipo genético y detectar pacientes en riesgo. La alternancia de la onda T (Figura 5) se define como la variación latido a latido de la amplitud, morfología y polaridad de la onda T en ritmo sinusal, sin variaciones en el complejo QRS. Constituye un indicador de inestabilidad eléctrica,50 refleja dispersión regional en la repolarización y en ocasiones precede a la fibrilación ventricular;51 puede ir acompañado de otros marcadores electrocardiográficos de alto riesgo como prolongación importante del QTc o bloqueo AV 2x1.

Bradicardia sinusal
Los pacientes con SQTL pueden cursar con signos de disfunción del nodo sinusal, bradicardia y/o pausas.52 Los subtipos SQTL1 y SQTL3, frecuentemente cursan con bradicardia sinusal,53 mientras el SQTL4 ha sido asociado a disfunción del nodo sinusal.32
Bloqueo AV 2x1
Desde los años 70–80's se observó la coexistencia de trastornos en la conducción aurículo–ventricular con el SQTL 54 (Figura 6). Es una manifestación infrecuente, pero de mal pronóstico, que puede presentarse desde la etapa fetal en forma de bradicardia persistente acompañada ocasionalmente de hidrops fetalis. La incidencia ha sido reportada entre 4 y 5%,55 y se asocia a una alta mortalidad a pesar del tratamiento con betabloqueadores y/o marcapaso.56,57 Este fenómeno puede explicarse por la exagerada duración del potencial de acción. Al alargarse el período refractario ventricular, el siguiente impulso procedente de la actividad sinoauricular, es bloqueado por encontrar a los ventrículos aún en periodo refractario. Así pues, el bloqueo 2x1 no se debe a una anormalidad intrínseca en la conducción AV, sino por una simple interferencia (bloqueo funcional). Esta alteración parece ser particular del SQTL, pues el periodo refractario ventricular es mayor que en el sistema de conducción AV.58 Hasta el momento tres genes han sido asociados con este grave fenotipo: HERG, (SQTL2),59,60 SCN5A (SQTL3)61 y CACNA1 (SQTL8).37

Taquicardia helicoidal
]]> Es la arritmia ventricular característica del SQTL, conocida también como "torsade de pointes"; se presenta cuando el intervalo QT se prolonga, independientemente de la etiología. Es una taquicardia ventricular polimórfica por reentrada, caracterizada electrocardiográficamente por un giro continuo del eje del QRS sobre una línea imaginaria (Figura 7); suele ser precedida de una pausa seguida de una extrasístole –intervalo RR "corto–largo–corto"– como se muestra en la figura 7.62–64 Puede culminar en fibrilación ventricular y muerte súbita. Si esto no sucede el paciente puede experimentar sólo un síncope, o incluso, si el episodio es breve, puede pasar inadvertido. Esta arritmia se explica mediante el fenómeno de dispersión de la refractariedad, según esta hipótesis, los periodos refractarios alargados de manera desproporcionada en diversas regiones ventriculares podrían alterar el patrón de conducción de las despolarizaciones ventriculares prematuras, causando bloqueo unidireccional y conducción lenta, lo cual dará lugar a un mecanismo de reentrada ventricular.65
Correlación genotipo–fenotipo en el ECG
Los diferentes genotipos pueden dar lugar a patrones electrocardiográficos característicos. Esto se ha evaluado en los subtipos más frecuentes, SQTL1, SQTL2 y SQTL3 (Figura 8). Los patrones son los siguientes:
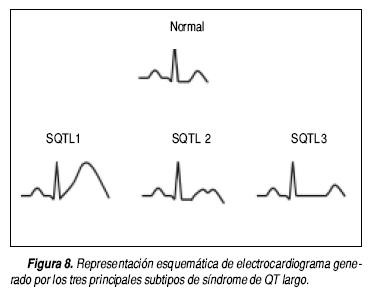
• SQTL1. Onda T de base ancha, con duración muy prolongada.
• SQTL2. Onda T de baja amplitud, bífida, con muescas.
]]> • SQTL 3. Onda T acuminada, de aparición tardía que deja observar con claridad el alargamiento del segmento ST.66
Diagnóstico prenatal de SQTL
La bradicardia fetal puede ser una de las primeras manifestaciones clínicas en el SQTL. En series retrospectivas, se ha documentado que hasta 70% de los pacientes diagnosticados en la infancia, tienen este antecedente, mismo que suele ir acompañado de hidrops fetalis.61 Mosaicismos para SQTL se han asociado a pérdidas fetales recurrentes durante el tercer trimestre del embarazo.68 Si la sospecha de la enfermedad es muy alta, la amniocentesis a partir de las 16 semanas de gestación puede ser de utilidad para el diagnóstico, que resulta sencillo cuando alguno de los padres es conocido como portador de una mutación determinada.69
Prolongación del intervalo QT y taquicardia helicoidal inducida por fármacos
La taquicardia helicoidal secundaria a fármacos no antiarrítmicos es un evento raro; se estima que ocurre menos de un caso por cada 10,000–100,000 expuestos. Considerando que los estudios clínicos incluyen entre 2,000 y 3,000 sujetos, es fácil que este indeseable y fatal efecto secundario escape a la detección como un efecto adverso en la fase clínica del desarrollo de fármacos.70 Este punto ha generado enorme interés en lo que se refiere a aspectos de seguridad en el estudio y generación de nuevas drogas.
En los últimos años se ha hecho evidente que existen gran variedad de fármacos utilizados en diversas especialidades médicas que pueden ocasionar el alargamiento del intervalo QT en forma iatrogénica. Incluso, algunos medicamentos han sido retirados del mercado en diferentes países por este indeseable efecto (p.ej. astemizol, cisaprida, entre otros). En el cuadro 3 se señalan algunos fármacos de uso frecuente que han sido asociados a prolongación del intervalo QT y taquicardia helicoidal.71,72 La lista continúa creciendo (para mayores detalles consultar el siguiente portal de internet: www.qtdrugs.org). Los factores relacionados con la susceptibilidad individual se ilustran en el cuadro 4.


El canal que por excelencia interacciona con fármacos es el IKr, codificado por el gen KCNH2 (HERG). Esto se debe a la estructura molecular de este canal. La mayoría de los canales que transportan el ion K+ tienen en el S6 de la α hélice dos residuos prolina que se inclinan en forma angulada hacia el poro del canal, disminuyendo su lumen, pero IKr carece de ellos, lo que hace que esta región del poro sea más amplia, facilitando su exposición a grandes moléculas. Por el contrario, tiene dos residuos aromáticos (tirosina y fenilalanina) que facilitan enlaces con moléculas aromáticas presentes en diversos fármacos capaces de bloquear el canal.73
Como mencionamos anteriormente, el SQTL tiene penetrancia incompleta, existen portadores asintomáticos de mutaciones que pueden manifestar arritmias malignas al recibir alguno de estos fármacos. Por otro lado, polimorfismos considerados frecuentes en la población, confieren susceptibilidad individual a desarrollar taquicardia helicoidal con el uso de fármacos, como sucede con el polimorfismo R1047L, el segundo más frecuentemente observado en KCNH2 (HERG) y que se ha asociado a taquicardia helicoidal con el uso de la droga dofetilide.74 Se han descrito por lo menos 20 polimorfismos en sujetos sanos en el gen KCNH2 o HERG y aún se ignora si confieren susceptibilidad individual a desarrollar taquicardia helicoidal relacionada con fármacos.75 En el canal de sodio Navl.5, también se han documentado polimorfismos que pueden conferir susceptibilidad a desarrollar arritmias ventriculares, como sucede con el polimorfismo H558R, que se presenta en cerca de 10–20% de la población; o el S1103Y, frecuente en afroamericanos76–80 y cuya implicación en la susceptibilidad a determinadas drogas no ha sido explorada.
Abordaje Inicial en el SQTL
Todo paciente que se encuentra en valoración por síncope o crisis convulsivas debe someterse a una meticulosa evaluación del intervalo QT. Una vez que se detecta un caso con SQTL, será necesario determinar el riesgo de muerte súbita del caso en particular, los siguientes elementos serán de utilidad para la estratificación del riesgo.
Historia clínica
Es crucial determinar si existen antecedentes familiares y/o personales de muerte súbita, así como el contexto del evento, que, como revisamos anteriormente, puede sugerirnos el subtipo de SQTL. Todos aquellos enfermos con antecedentes familiares de muerte súbita, tienen mayor probabilidad de presentar arritmias ventriculares malignas. Es importante descartar el uso de fármacos que pudieran prologar el QT y dar así "falsos positivos" en la evaluación electrocardiográfica inicial.
]]>Holter
El monitoreo Holter permite una valoración amplia y dinámica del intervalo QT; en ocasiones, pueden registrarse episodios espontáneos de taquicardia helicoidal asintomática y los factores precipitantes de ésta, así como posibles episodios de bloqueos AV.
Prueba de esfuerzo
Los pacientes con SQTL no suelen alcanzar la frecuencia máxima esperada calculada para la edad, así mismo, el intervalo QT al esfuerzo puede tener un comportamiento paradójico, alargándose en lugar de acortarse.81,82 El comportamiento electrocardiográfico durante la prueba de esfuerzo, será diferente según el subtipo SQTL. Los pacientes con SQTL1 además de no llegar a la frecuencia cardiaca máxima calculada para la edad, frecuentemente alargan el intervalo QT, mientras que aquellos con SQTL2, suelen alcanzar la frecuencia cardiaca esperada, y prolongar sólo discretamente el intervalo QT, o incluso no prolongarlo.83,84 Los pacientes con SQTL3 normalmente tienen una respuesta fisiológica al ejercicio, esto es, acortamiento normal del intervalo QT.85 Este estudio también puede ser útil para valorar la respuesta al tratamiento y estratificar el riesgo en los casos asintomáticos o en quienes queda duda de los factores precipitantes de las arritmias.
Estudio genético
El diagnóstico genético–molecular es una herramienta extremadamente útil en el manejo de los pacientes con SQTL.86 Su principal aplicación será el consejo genético, pero también permite orientar el tratamiento y es útil en la evaluación del pronóstico. Si bien se utiliza cada vez con mayor frecuencia, en la gran mayoría de los países aún se limita a hospitales que cuentan con laboratorios de investigación. Con los conocimientos actuales, la probabilidad de diagnosticar genéticamente a un paciente determinado es de 70–75%, en 25–30% no es posible documentar mutaciones en los genes descritos a la fecha,13'87 esto tiene varias explicaciones:
1. Otros genes, no descritos aún, pudieran estar asociados a la enfermedad. Los canales funcionan en complejos macromoleculares, diversas proteínas son cruciales en su funcionamiento y el trastorno puede encontrarse no en el canal en sí, sino en las proteínas que interactúan con él, como ya se demostró con las mutaciones en Ankirina β y las subunidades auxiliares β de los canales de potasio IKr e IKs.
]]> 2. El problema puede radicar en el procesamiento del ARNm, no detectable en el análisis convencional de ADN extraído de linfocitos.3. Se han reportado mutaciones intrónicas asociadas a SQTL, en el estudio rutinario no se analizan estas regiones.88
4. Algunos mosaicismos para SQTL pueden no ser detectados en el estudio de ADN extraído de linfocitos.68
El SQTL es una enfermedad de presentación principalmente monogénica,89 las variedades poligénicas o compuestas pueden dar un fenotipo más grave. La penetrancia es muy variable, oscilando entre 25 y 90%.90 Hasta 32% de los portadores de la mutación pueden cursar con un QTc normal. Estos portadores asintomáticos tienen un riesgo de 50% de transmitir la mutación a su descendencia y son más susceptibles a desarrollar arritmias malignas, comparados con el resto de la población. De hecho, 20% de estos casos pueden volverse sintomáticos a pesar de tener un QTc dentro de los límites normales. Las manifestaciones de la enfermedad son variables, pues una misma mutación puede dar fenotipos diferentes o bien características combinadas, como serían el bloqueo AV y la prolongación del intervalo QT; o bien trastornos de la conducción intraventricular y QT largo. Es un padecimiento con gran heterogeneidad genética, pues hasta ahora ocho genes distintos pueden dar el mismo fenotipo (prolongación del intervalo QT), y las mutaciones en estos genes han explicado sólo 70–75% del total de los casos. A pesar de que el fenotipo final, esto es, prolongación del intervalo QT es igual para todas las mutaciones; la fisiopatología molecular es diferente y esto determina el curso clínico, es razonable entonces otorgar tratamiento dirigido al subtipo molecular.91 Por ejemplo: se ha documentado que pacientes con mutaciones en el poro del canal IKr codificado por HERG, tienen peor pronóstico, comparado con aquellos que tienen mutaciones en otros sitios del canal.92 Asimismo, los pacientes diagnosticados con SQTL3 pueden mejorar significativamente al recibir tratamiento con mexiletina93 (ver más adelante).
Reto farmacológico con adrenalina (epinefrina)
Algunos grupos han empleado el reto farmacológico con adrenalina para desenmascarar los casos sospechosos de SQTL con un QTc limítrofe. Este método ha sido particularmente útil en los subtipos SQTL1 y SQTL2, que suelen presentar una respuesta paradójica a la administración de dosis bajas de adrenalina (0.025–0.2 µg/kg/min), manifestando un alargamiento del intervalo QTc, cuando la respuesta normal es un acortamiento de dicho intervalo.94–96
Estudio electrofisiológico
No existe indicación de estudio electrofisiológico en pacientes con SQTL, a menos que se sospeche la presencia de alguna otra arritmia o bien como parte del protocolo de implante de desfibrilador.
]]>Estratificación de riesgo
La evolución de los casos con SQTL es muy variable, siendo influenciada por la duración del intervalo QTc, factores ambientales, edad, genotipo y respuesta al tratamiento.97 Se sabe que el riesgo de presentar eventos cardíacos es mayor en hombres antes de la pubertad y en mujeres en la vida adulta.98 Asimismo, las arritmias ventriculares son más frecuentes en SQTL1 y SQTL2, pero son más letales en SQTL3.99 Las mujeres en posparto son particulamiente susceptibles a arritmias malignas.100 Por otro lado, como ya mencionamos, las mutaciones en el poro del canal codificado por HERG, determinan presentaciones más graves de la enfermedad.
Riesgo alto
En términos generales, debe considerarse de alto riesgo aquellos casos de SQTL asociado a:
1. Sordera congénita (síndrome de Jervell–Lange–Nielsen).
2. Síncope recurrente por taquiarritmias ventriculares malignas.
a) Antecedentes familiares de muerte súbita.
b) QTc > 500 ms.
]]> 3. Bloqueo AV 2x1.4. Alternancia eléctrica en la onda T.
Los estudios realizados por Priori, et al., en 647 pacientes, mostraron que la probabilidad de presentar un evento mayor (síncope, paro cardíaco, muerte súbita) antes de los 40 años es alto (> 50%) cuando el QTc > 500 mseg en SQTL1, SQTL2 o bien en el sexo masculino cuando existe SQTL3.97
Riesgo intermedio
La posibilidad de presentar un evento mayor es intermedio (30–49%) cuando QTc < 500 mseg en mujeres con SQTL2, en SQTL3, o bien en mujeres con SQTL3 y QTc > 500 mseg.
Riesgo bajo
La posibilidad de presentar un evento mayor es bajo (< 30%) en aquellos pacientes con QTc < 500 mseg en hombres con SQTL2 y SQTL1. Los pacientes asintomáticos con intervalo QTc prolongado y sin historia familiar de muerte súbita tienen una baja incidencia de crisis de taquiarritmias, así como aquellos con QTc prolongado intermitente o limítrofe.
Dada su baja incidencia, no hay estudios que permitan clasificar el riesgo en los otros subtipos de SQTL.
]]>Síndrome de QT largo y embarazo
Durante el embarazo existen cambios fisiológicos importantes que pueden favorecer arritmias, sobre todo durante el parto: ajustes hormonales, en particular incremento en hormonas tiroideas, cambios en el tono autonómico, hipokalemia, entre otras. Las pacientes portadoras de SQTL son más susceptibles a arritmias malignas en el periodo posparto, por lo que la vigilancia estrecha en este periodo es importante.101 El SQTL2 (KCNH2 o HERG) es el que con mayor frecuencia presenta arritmias durante el posparto.100
Estrategias terapéuticas en el SQTL
Los pacientes que no reciben tratamiento tienen una mortalidad de 20% al año y de 50% a 10 años después de un primer evento de arritmia ventricular. En la actualidad el tratamiento de elección depende del subtipo de SQTL y de los síntomas del paciente. La restricción en el ejercicio será siempre recomendable, sobre todo en los casos de riesgo moderado a alto.
FÁRMACOS
Betabloqueadores
Los betabloqueadores son eficaces, en particular, en pacientes con mutaciones en canal de potasio (SQTL1, SQTL2, SQTL5 y SQTL6). Los resultados son mejores en aquellos pacientes con mutaciones en IKs (SQTL1, SQTL5)102 que, como se describió anteriormente, es regulado por el sistema simpático. Los betabloqueadores no modifican el intervalo QT, pero son útiles en prevenir las arritmias ventriculares. En pacientes con SQTL3, no han mostrado beneficio importante y de hecho, deberán usarse con cautela, pues los episodios de arritmia ventricular en este subtipo son más comunes a frecuencias cardiacas bajas. De preferencia, los pacientes con SQTL3 deberán contar con un marcapaso antes del inicio del tratamiento. Se ha demostrado que si bien los betabloqueadores disminuyen la incidencia de eventos cardiovasculares,103 no garantizan protección contra la muerte súbita; a pesar del tratamiento, 19% de los pacientes con SQTL1, y 41% con SQTL2, tendrán recurrencia de los síntomas.104,105
]]> Para establecer la dosis adecuada, es útil la prueba de esfuerzo. La frecuencia cardiaca máxima no debe superar los 130 lpm, bajo tratamiento.
Bloqueadores de canales de sodio
Dado que las mutaciones en el canal de sodio que ocasionan el SQTL3 se deben a una inactivación defectuosa del canal, se han utilizado bloqueadores de canales de sodio en pacientes con diagnóstico genético concluyente. Estudios realizados con flecainida muestran que mejora la frecuencia cardiaca, las alteraciones en la onda T y el intervalo QT.106 La mexiletina también ha mostrado mejorar los marcadores electrocardiográficos de riesgo.85,107,108 Si bien los resultados son alentadores, hay que considerar que no existe un seguimiento a largo plazo que evalúe la evolución de los enfermos tratados con estos fármacos, ni se han utilizado en un número importante de pacientes. No se deben administrar si no existe una certeza en el diagnóstico genético y su administración inicial debe ser monitorizada en forma intrahospitalaria.
Potasio suplementario y fármacos que facilitan su disponibilidad
En aquellos enfermos con afección en canales de potasio, el suplemento de potasio y/o la utilización de fármacos ahorradores de potasio como espironolactona, pueden disminuir el intervalo QTc hasta en 24%.109,110 Los fármacos que abren los canales de potasio como apricalium, leveromakalium, nicorandil y pinacidil, han mostrado ser útiles,111 pero aún no se encuentran disponibles en nuestro medio.
Marcapaso
La implantación de un marcapaso está indicado en todos aquellos pacientes en los que se documente o se sospeche arritmia dependiente de pausa.112 Los pacientes con SQTL3 suelen beneficiarse más de esta terapéutica, pues la prevalencia de bradicardia es mayor. En el SQTL2 los eventos cardiovasculares pueden presentarse durante la noche, por lo que también se consideran candidatos. Si bien en el SQTL1 el tratamiento angular son los betabloqueadores, algunos casos pueden beneficiarse con el implante de un marcapaso. La programación adecuada del dispositivo es de crucial importancia:
]]>Frecuencia cardiaca
Está demostrado que frecuencias programadas por debajo de 70 no son útiles para prevenir arritmias ventriculares.113 En adultos se recomienda programar una FC promedio de 80 1pm, aunque no deberá exceder los 90 1pm en reposo.
Modalidad de estimulación
La estimulación debe ser DDD en los pacientes que presenten taquicardia helicoidal dependiente de pausa, también será la modalidad de elección en los casos con bloqueo AV 2x1 o de alto grado.
Sensor
Se recomienda programar el sensor a una respuesta rápida, pues estos pacientes suelen tener una aceleración inapropiada de la frecuencia cardiaca en respuesta al ejercicio.
]]> Otras funciones
Todas aquellas funciones que impliquen la presencia de pausas, deben ser apagadas. Se debe apagar la histéresis, la función nocturna (disminución de frecuencia cardiaca durante el sueño) y el alargamiento del PVARP seguido de extrasístole ventricular, pues si bien previene la existencia de taquicardia en asa, da lugar a pausas. Si existe la función de regulación de frecuencia (rate–smoothing), debe ser encendida para prevenir pausa postextrasistólica. Hay que evitar el sobresensado de la onda T o fallas en la captura, que pueden dar lugar a pausas.
Desfibrilador
El desfibrilador (DAI) es recomendable en aquellos pacientes catalogados como de alto riesgo de muerte súbita. En este grupo de enfermos, el desfibrilador aunado a terapia con betabloqueador, disminuye en forma importante la incidencia de muerte súbita.114–116 La programación del dispositivo variará según las necesidades del caso, pero se sugiere prestar atención a los siguientes aspectos:
Rango de detección
Es importante definir a qué frecuencia ventricular el aparato deberá administrar una descarga, el rango de detección debe ser lo suficientemente bajo para no omitir tratamientos sin generar terapias inapropiadas. Se ha sugerido que en estos casos, una frecuencia de detección a 180 lpm (zona de TV) con un prolongado tiempo de detección de la arritmia, evitará eventos de arritmia ventricular no tratados. Para frecuencias mayores de 210 lpm (zona de FV), se puede disminuir el tiempo de detección.114
Tormenta arrítmica
]]> Cerca de 15% de los pacientes pueden presentar tormenta arrítmica por el incremento del tono simpático después de la descarga del DAI; este problema puede manejarse inicialmente incrementando la terapia con betabloqueadores y con terapia antibradicardia con el marcapaso; en los casos graves que no responden a estas medidas, se debe considerar realizar resección de la cadena ganglionar simpática, como se describirá más adelante.
Terapias inapropiadas
Se ha observado que frecuentemente los pacientes con SQTL presentan terapias inapropiadas en respuesta a taquicardia sinusal o arritmias auriculares. El tratamiento con betabloqueadores ayudará a disminuir estos episodios. Por otro lado, hay que evitar dar terapia eléctrica a los episodios breves de arritmia ventricular, especialmente en pacientes con múltiples episodios de corta duración y pocos síntomas; la prolongación de los tiempos de detección de la arritmia disminuirá el número de descargas.
Simpatectomía izquierda
Desde 1971, se introdujo la gangliectomía simpática como opción terapéutica útil en estos pacientes.117 En 1991, Schwartz publicó la primera serie de 85 casos con pobre respuesta al tratamiento betabloqueador, en los que se realizó estelectomía izquierda con resultados alentadores, pues se logró una sobrevida a cinco años de 94%.118 Actualmente se ofrece esta opción terapéutica a pacientes de alto riesgo, cuando a pesar del tratamiento con betabloqueador y/o marcapaso el paciente persiste con síncope, o bien si una vez implantado el desfibrilador, existen descargas frecuentes.
El procedimiento consiste en realizar la resección de la porción inferior del ganglio estrellado y de las cadenas ganglionares torácicas simpáticas izquierdas T2 a T4, ya que la simple estelectomía izquierda ha mostrado no ser suficientemente efectiva. Se ha utilizado la toracoscopia microinvasiva con buenos resultados.119,120 Recientemente se publicó la serie más importante de pacientes tratados con este método, la cual mostró una reducción significativa en los episodios de síncope o muerte súbita, así como con una sobrevida a cinco años de 95%. En pacientes con síncope previo, la sobrevida a cinco años fue de 97% con 11% de posibilidades de recurrencia, los cuales fueron en la mayoría un solo evento sincopal aislado. Asimismo, se encontró una reducción significativa del segmento QT posterior a la simpatectomía izquierda. A pesar de los buenos resultados, la prevención de muerte súbita no es completa, pero se logra reducir a 3%. En pacientes portadores de un desfibrilador automático implantable en los que se realizó la cirugía por la presencia de múltiples descargas, la media de eventos fue de 25 a 0 con una reducción de 95%. En el SQTL1, se confirmó el efecto benéfico. Probablemente el beneficio sea menor en pacientes con SQTL2. Hasta ahora no se ha logrado establecer su efectividad para el SQTL3.121
Ablación
]]> Se ha reportado que es posible realizar ablación de la extrasístole que en algunos casos inicia la arritmia ventricular, con mejoría en la incidencia de episodios de taquicardia helicoidal.122 Sin embargo, no existen estudios a largo plazo ni con un número de pacientes apropiado que justifique utilizar esta técnica en forma rutinaria. Probablemente sea de utilidad como parte del tratamiento en algunos casos en particular, como en pacientes con tormenta arrítmica y descargas frecuentes del desfibrilador.
Autopsia molecular
Se propone como alternativa en los casos con muerte súbita en quienes la autopsia no explica en forma clara la causa de muerte.123 El estudio genético posmortem se ha utilizado con anterioridad y en forma interesante se han encontrado mutaciones que condicionan SQTL en porcentajes variables.124,125 Este tipo de estudios, además de poder tener repercusiones legales, tiene implicaciones en el consejo genético a los familiares de los casos que pudieran estar afectados sin saberlo.
Por medio de la autopsia molecular se ha podido concluir que las canalopatías arritmogénicas pueden ser otra causa más de la "muerte de cuna"; en 5–10% de los casos se han encontrado mutaciones en los genes relacionados sólo al SQTL,126 sin mencionar otras canalopatías.
AGRADECIMIENTOS
La Dra. Medeiros recibe apoyo económico de CONACyT y FUNSALUD.
REFERENCIAS
]]>1. Wever EF, Robles de Medina EO. Sudden death in patients without structural heart disease. J Am Coll Cardiol 2004; 43: 1137–44. [ Links ]
2. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998; 98: 2334–51. [ Links ]
3. Chugh SS, Kelly KL, Titus JL. Sudden cardiac death with apparently normal heart. Circulation. 2000; 102: 649–54. [ Links ]
4. Puranik R, Chow CK, Duflou JA, Kilborn MJ, McGuire MA. Sudden death in the young. Heart Rhythm. 2005; 2: 1277–82. [ Links ]
5. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005; 85: 1205–53. [ Links ]
6. Herrlin KMMT. A case of cardiac syncope. Acta Paediatriva 1953; 391. [ Links ]
7. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995; 80: 795–803. [ Links ]
8. Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 1995; 4: 1603–7. [ Links ]
9. Beccaria E, Brim S, Gaita F, Giustetto C, Conti M. Torsade de pointes during an atropine sulfate test in a patient with congenital long QT syndrome. Cardiología 1989; 34: 1039–43. [ Links ]
10. Jervell A, Lange–Nielsen F. Congenital deaf–mutism, functional heart disease with prolongation of the Q–T interval and sudden death. Am Heart J 1957; 54: 59–68. [ Links ]
11. Romano C, Gemme G, Pongiglione R. Rare cardiac arrhythmias of the pediatric age. I. Repetitive Paroxysmal Tachycardia. Minerva Pediatr 1963; 15: 1155–64. [ Links ]
12. Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995; 80: 805–11. [ Links ]
13. Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005; 294: 2975–80. [ Links ]
14. Nerbonne JM. Molecular basis of functional voltage–gated K+ channel diversity in the mammalian myocardium. J Physiol 2000; 525 Pt 2: 285–98. [ Links ]
15. Crozier IG, Loughnan A, Dow LJ, Low CJ, Ikram H. Congenital long QT syndrome in adults. N Z Med J 1989; 102: 340–1. [ Links ]
16. McCrossan ZA, Abbott GW. The MinK–related peptides. Neuropharmacology 2004; 47: 787–821. [ Links ]
17. Abbott GW, Goldstein SA. Potassium channel subunits encoded by the KCNE gene family: physiology and pathophysiology of the MinK–related peptides (MiRPs). Mol Interv 2001; 1: 95–107. [ Links ]
18. Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1–KCNE1 potassium channel. Science 2002; 295: 496–9. [ Links ]
19. Schwartz PJ. Another role for the sympathetic nervous system in the long QT syndrome? J Cardiovasc Electrophysiol 2001; 12: 500–2. [ Links ]
20. Goldin AL, Barchi RL, Caldwell JH, et al. Nomenclature of voltage–gated sodium channels. Neuron 2000; 28: 365–8. [ Links ]
21. Gellens ME, George AL, Jr., Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin–insensitive voltage–dependent sodium channel. Proc Nati Acad Sci USA 1992; 89: 554–8. [ Links ]
22. Ko SH, Lenkowski PW, Lee HC, Mounsey JP, Patel MK. Modulation of Na(v)1.5 by betal— and beta3–subunit co–expression in mammalian cells. Pflugers Arch 2005; 449: 403–12. [ Links ]
23. Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel alpha and beta subunits in single ventricular myocytes from mouse heart. Circulation 2004; 109: 1421–7. [ Links ]
24. Wollner DA, Messner DJ, Catterall WA. Beta 2 subunits of sodium channels from vertebrate brain. Studies with subunit–specific antibodies. J Biol Chem 1987; 262: 14709–15. [ Links ]
25. Isom LL, Ragsdale DS, De Jongh KS, et al. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 1995; 83: 433–42. [ Links ]
26. Morgan K, Stevens EB, Shah B, et al. Beta 3: an additional auxiliary subunit of the voltage–sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Nati Acad Sci USA 2000; 97: 2308–13. [ Links ]
27. Yu FH, Westenbroek RE, Silos–Santiago I, et al. Sodium channel beta4, a new disulfide–linked auxiliary subunit with similarity to beta2. J Neuro sci 2003; 23: 7577–85. [ Links ]
28. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype–phenotype correlation in the long–QT syndrome: gene–specific triggers for life–threatening arrhythmias. Circulation 2001; 103: 89–95. [ Links ]
29. Beaufort–Krol GC, van den Berg MP, Wilde AA, et al. Developmental aspects of long QT syndrome type 3 and Brugada syndrome on the basis of a single SCN5A mutation in childhood. J Am Coll Cardiol 2005; 46: 331–7. [ Links ]
30. Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin–B mutation causes type 4 long–QT cardiac arrhythmia and sudden cardiac death. Nature 2003; 421: 634–9. [ Links ]
31. Mohler PJ, Bennett V. Ankyrin–based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol 2005; 20: 189–93. [ Links ]
32. Mohler PJ, Splawski I, Napolitano C, et al. A cardiac arrhythmia syndrome caused by loss of ankyrin–B function. Proc Nati Acad Sci USA 2004; 101: 9137–42. [ Links ]
33. Splawski I, Tristani–Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 1997; 17: 338–40. [ Links ]
34. Abbott GW, Sesti F, Splawski I, et al. MiRPl forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999; 97: 175–87. [ Links ]
35. Plaster NM, Tawil R, Tristani–Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001; 105: 511–19. [ Links ]
36. Yoon G, Oberoi S, Tristani–Firouzi M, et al. Andersen–Tawil syndrome: Prospective cohort analysis and expansion of the phenotype. Am J Med Genet A 2006; 140: 312–21. [ Links ]
37. Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19–31. [ Links ]
38. Splawski I, Timothy KW, Decher N, et al. Severe arrhythmia disorder caused by cardiac L–type calcium channel mutations. Proc Nati Acad Sci USA 2005; 102: 8089–96; discussion 8086–8. [ Links ]
39. Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long–QT syndrome. Circulation 2004; 109: 1834–41. [ Links ]
40. Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange–Nielsen Syndrome. Natural History, Molecular Basis, and Clinical Outcome. Circulation 2006. [ Links ]
41. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88: 782–4. [ Links ]
42. Viskin S, Rosovski U, Sands AJ, et al. Inaccurate electrocardiographic interpretation of long QT: the majority of physicians cannot recognize a long QT when they see one. Heart Rhythm 2005; 2: 569–74. [ Links ]
43. Cowan JC, Yusoff K, Moore M, et al. Importance of lead selection in QT interval measurement. Am J Cardiol 1988; 61: 83–7. [ Links ]
44. Brugada RHK, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, et al. Sudden death associated with short–QT syndrome linked to mutations in HERG. Circulation 2004; 109: 30–35. [ Links ]
45. Antzelevitch C, Belardinelli L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol 2006; 17 (Suppl. 1): S79–S85. [ Links ]
46. Yamaguchi M, Shimizu M, Ino H, et al. T wave peak–to–end interval and QT dispersion in acquired long QT syndrome: a new index for arrhythmogenicity. Clin Sci (Lond) 2003; 105: 671–6. [ Links ]
47. Priori SG, Napolitano C, Diehl L, Schwartz PJ. Dispersion of the QT interval. A marker of therapeutic efficacy in the idiopathic long QT syndrome. Circulation 1994; 89: 1681–9. [ Links ]
48. Napolitano C, Priori SG, Schwartz PJ. Significance of QT dispersion in the long QT syndrome. Prog Cardiovasc Dis 2000; 42: 345–50. [ Links ]
49. Perkiomaki JS, Zareba W, Nomura A, Andrews M, Kaufman ES, Moss AJ. Repolarization dynamics in patients with long QT syndrome. J Cardiovasc Electrophysiol 2002; 13: 651–6. [ Links ]
50. Zareba W, Moss AJ, le Cessie S, Hall WJ. T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol 1994; 23: 1541–6. [ Links ]
51. Narayan SM. T–wave alternans and the susceptibility to ventricular arrhythmias. J Am Coll Cardiol 2006; 47: 269–81. [ Links ]
52. Beinder E, Grancay T, Menendez T, Singer H, Hofbeck M. Fetal sinus bradycardia and the long QT syndrome. Am J Obstet Gynecol 2001; 185: 743–7. [ Links ]
53. Lupoglazoff JM, Denjoy I, Villain E, et al. Long QT syndrome in neonates: conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol 2004; 43: 826–30. [ Links ]
54. Scott WA, Dick M, 2nd. Two:one atrioventricular block in infants with congenital long QT syndrome. Am J Cardiol 1987; 60: 1409–10. [ Links ]
55. Garson A Jr., Dick M, 2nd, Fournier A, et al. The long QT syndrome in children. An international study of 287 patients. Circulation 1993; 87: 1866–72. [ Links ]
56. Trippel DL, Parsons MK, Gillette PC. Infants with long–QT syndrome and 2:1 atrioventricular block. Am Heart J 1995; 130: 1130–4. [ Links ]
57. Gorgels AP, Al Fadley F, Zaman L, Kantoch MJ, Al Halees Z. The long QT syndrome with impaired atrioventricular conduction: a malignant variant in infants. J Cardiovasc Electrophysiol 1998; 9: 1225–32. [ Links ]
58. van Hare GF, Franz MR, Rogé C, Scheinman MM. Persistent functional atrioventricular block in two patients with prolonged QT intervals: elucidation of the mechanism of block. Pacing Clin Electrophysiol 1990; 13: 608–18. [ Links ]
59. Hoorntje T, Alders M, van Tintelen P, et al. Homozygous premature truncation of the HERG protein: the human HERG knockout. Circulation 1999; 100: 1264–7. [ Links ]
60. Pilippo K, Laitinen P, Swan H, et al. Homozygosity for a HERG potassium channel mutation causes a severe form of long QT syndrome: identification of an apparent founder mutation in the Finns. J Am Coll Cardiol 2000; 35: 1919–25. [ Links ]
61. Lupoglazoff JM, Cheav T, Baroudi G, et al. Homozygous SCN5A mutation in long–QT syndrome with functional two–to–one atrioventricular block. Circ Res 2001; 89: E16–E21. [ Links ]
62. Noda T, Shimizu W, Satomi K, et al. Classification and mechanism of Torsade de Pointes initiation in patients with congenital long QT syndrome. Eur Heart J 2004; 25: 2149–54. [ Links ]
63. Liu J, Laurita KR. The mechanism of pause–induced torsade de pointes in long QT syndrome. J Cardiovasc Electrophysiol 2005; 16: 981–7. [ Links ]
64. Viskin S, Fish R, Zeltser D, et al. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart 2000; 83: 661–6. [ Links ]
65. el–Sherif N, Turitto G. The long QT syndrome and torsade de pointes. Pacing Clin Electrophysiol 1999; 22: 91–110. [ Links ]
66. Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST–T–wave patterns and repolarization parameters in congenital long–QT syndrome: ECG findings identify genotypes. Circulation 2000; 102: 2849–55. [ Links ]
67. Chang IK, Shyu MK, Lee CN, et al. Prenatal diagnosis and treatment of fetal long QT syndrome: a case report. Prenat Diagn 2002; 22: 1209–12. [ Links ]
68. Miller TE, Estrella E, Myerburg RJ, et al. Recurrent third–trimester fetal loss and maternal mosaicism for long–QT syndrome. Circulation 2004; 109: 3029–34. [ Links ]
69. Tester DJ, McCormack J, Ackerman MJ. Prenatal molecular genetic diagnosis of congenital long QT syndrome by strategic genotyping. Am J Cardiol 2004; 93: 788–91. [ Links ]
70. Fitzgerald PT, Ackerman MJ. Drug–induced torsades de pointes: the evolving role of pharmacogenetics. Heart Rhythm 2005; 2: S30–7. [ Links ]
71. Roden DM. Drug–induced prolongation of the QT interval. N Engl J Med 2004; 350: 1013–22. [ Links ]
72. Viskin S, Justo D, Halkin A, Zeltser D. Long QT syndrome caused by noncardiac drugs. Prog Cardiovasc Dis 2003; 45:415–27. [ Links ]
73. Abriel H, Schlapfer J, Keller DI, et al. Molecular and clinical determinants of drug–induced long QT syndrome: an iatrogenic channelopathy. Swiss Med Wkly 2004; 134: 685–94. [ Links ]
74. Sun Z, Milos PM, Thompson JF, et al. Role of a KCNH2 polymorphism (R1047 L) in dofetilide–induced Torsades de Pointes. J Mol Cell Cardiol 2004; 37: 1031–9. [ Links ]
75. Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc 2003; 78: 1479–87. [ Links ]
76. Ackerman MJ, Splawski I, Makielski JC, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenie susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004; 1: 600–7. [ Links ]
77. Ye B, Valdivia CR, Ackerman MJ, Makielski JC. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics 2003; 12: 187–93. [ Links ]
78. Makielski JC, Ye B, Valdivia CR, et al. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res 2003; 93: 821–8. [ Links ]
79. Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med 2006; 259: 59–69. [ Links ]
80. Plant LD, Bowers PN, Liu Q, et al. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J Clin Invest 2006; 116: 430–5. [ Links ]
81. Walker BD, Krahn AD, Klein GJ, Skanes AC, Yee R. Burst bicycle exercise facilitates diagnosis of latent long QT syndrome. Am Heart J 2005; 150: 1059–63. [ Links ]
82. Vincent GM, Jaiswal D, Timothy KW. Effects of exercise on heart rate, QT, QTc and QT/QS2 in the Romano–Ward inherited long QT syndrome. Am J Cardiol 1991; 68: 498–503. [ Links ]
83. Takenaka K, Ai T, Shimizu W, et al. Exercise stress test amplifies genotype–phenotype correlation in the LQT1 and LQT2 forms of the long–QT syndrome. Circulation 2003; 107: 838–44. [ Links ]
84. Swan H, Viitasalo M, Piippo K, Laitinen P, Kontula K, Toivonen L. Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and HERG potassium channel defects. J Am Coll Cardiol 1999; 34: 823–9. [ Links ]
85. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene–specific therapy. Circulation 1995; 92: 3381–6. [ Links ]
86. Schwartz PJ. Management of long QT syndrome. Nat Clin Pract Cardiovasc Med 2005; 2: 346–51. [ Links ]
87. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005; 2: 507–17. [ Links ]
88. Zhang L, Vincent GM, Baralle M, et al. An intronic mutation causes long QT syndrome. J Am Coll Cardiol 2004; 44: 1283–91. [ Links ]
89. Priori SG. Inherited arrhythmogenic diseases: the complexity beyond monogenic disorders. Circ Res 2004; 94: 140–5. [ Links ]
90. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long–QT syndrome: clinical impact. Circulation 1999; 99: 529–33. [ Links ]
91. Schwartz PJ. Clinical applicability of molecular biology: the case of the long QT syndrome. Curr Control Trials Cardiovasc Med 2000; 1: 88–91. [ Links ]
92. Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long–QT syndrome with mutations in the pore region of the human ether–a–go–go–related gene potassium channel. Circulation 2002; 105: 794–9. [ Links ]
93. Yao CT, Wang JN, Tsai YC, Lin CS, Wu JM. Congenital long QT syndrome with functionally impaired atrioventricular conduction: successful treatment by mexiletine and propranolol. J Formos Med Assoc 2002; 101: 291–3. [ Links ]
94. Ackerman MJ, Khositseth A, Tester DJ, Hejlik JB, Shen WK, Porter CB. Epinephrine–induced QT interval prolongation: a gene–specific paradoxical response in congenital long QT syndrome. Mayo Clin Proc 2002; 77: 413–21. [ Links ]
95. Khositseth A, Hejlik J, Shen WK, Ackerman MJ. Epinephrine–induced T–wave notching in congenital long QT syndrome. Heart Rhythm 2005; 2: 141–6. [ Links ]
96. Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm 2004; 1: 276–83 [ Links ].
97. Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long–QT syndrome. N Engl J Med 2003; 348: 1866–74. [ Links ]
98. Locati EH, Zareba W, Moss AJ, et al. Age–and sex–related differences in clinical manifestations in patients with congenital long–QT syndrome: findings from the International LQTS Registry. Circulation 1998; 97: 2237–44. [ Links ]
99. Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long–QT syndrome. International Long–QT Syndrome Registry Research Group. N Engl J Med 1998; 339: 960–5. [ Links ]
100. Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm 2004; 1: 60–4. [ Links ]
101. Rashba EJ, Zareba W, Moss AJ, et al. Influence of pregnancy on the risk for cardiac events in patients with hereditary long QT syndrome. LQTS Investigators. Circulation 1998; 97: 451–6. [ Links ]
102. Itoh T, Kikuchi K, Odagawa Y, et al. Correlation of genetic etiology with response to beta–adrenergic blockade among symptomatic patients with familial long–QT syndrome. J Hum Genet 2001; 46: 38–40. [ Links ]
103. Chatrath R, Bell CM, Ackerman MJ. Beta–blocker therapy failures in symptomatic probands with genotyped long–QT syndrome. Pediatr Cardiol 2004; 25: 459–65. [ Links ]
104. Dorostkar PC, Eldar M, Belhassen B, Scheinman MM. Long–term follow–up of patients with long–QT syndrome treated with beta–blockers and continuous pacing. Circulation 1999; 100: 2431–6. [ Links ]
105. Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta–blockers. JAMA 2004; 292: 1341–4. [ Links ]
106. Benhorin J, Taub R, Goldmit M, et al. Effects of flecainide in patients with new SCN5A mutation: mutation–specific therapy for long–QT syndrome? Circulation 2000; 101: 1698–706. [ Links ]
107. Schulze–Bahr E, Fenge H, Etzrodt D, et al. Long QT syndrome and life threatening arrhythmia in a newborn: molecular diagnosis and treatment response. Heart 2004; 90: 13–6. [ Links ]
108. Kehl HG, Haverkamp W, Rellensmann G, et al. Images in cardiovascular medicine. Life–threatening neonatal arrhythmia: successful treatment and confirmation of clinically suspected extreme long QT–syndrome–3. Circulation 2004; 109: e205–e206. [ Links ]
109. Shimizu W, Kurita T, Matsuo K, et al. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long–QT syndrome. Circulation 1998; 97: 1581–8. [ Links ]
110. Etheridge SP, Compton SJ, Tristani–Firouzi M, Mason JW. A new oral therapy for long QT syndrome: long–term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol 2003; 42: 1777–82. [ Links ]
111. Khan IA, Gowda RM. Novel therapeutics for treatment of long–QT syndrome and torsade de pointes. Int J Cardiol 2004; 95: 1–6. [ Links ]
112. Viskin S. Cardiac pacing in the long QT syndrome: review of available data and practical recommendations. J Cardiovasc Electrophysiol 2000; 11: 593–600. [ Links ]
113. Moss AJ, Liu JE, Gottlieb S, Locati EH, Schwartz PJ, Robinson JL. Efficacy of permanent pacing in the management of high–risk patients with long QT syndrome. Circulation 1991; 84: 1524–9. [ Links ]
114. Monnig G, Kobe J, Loher A, et al. Implantable cardioverter–defibrillator therapy in patients with congenital long–QT syndrome: a long–term follow–up. Heart Rhythm 2005; 2: 497–504. [ Links ]
115. Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high–risk long QT syndrome patients. J Cardiovasc Electrophysiol 2003; 14: 337–41. [ Links ]
116. Kaufman ES. Saving lives in congenital long QT syndrome: who benefits from implantable cardioverter defibrillator therapy? J Cardiovasc Electrophysiol 2003; 14: 342–3. [ Links ]
117. Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N Engl J Med 1971; 285: 903–4. [ Links ]
118. Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation 1991; 84: 503–11. [ Links ]
119. Wang LX. Role of left cardiac sympathetic denervation in the management of congenital long QT syndrome. J Postgrad Med 2003; 49: 179–81. [ Links ]
120. Wang L, Feng G. Left cardiac sympathetic denervation as the first–line therapy for congenital long QT syndrome. Med Hypotheses 2004; 63: 438–41. [ Links ]
121. Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high–risk patients affected by the long–QT syndrome. Circulation 2004; 109: 1826–33. [ Links ]
122. Haissaguerre M, Extramiana F, Hocini M, et al. Mapping and ablation of ventricular fibrillation associated with long–QT and Brugada syndromes. Circulation 2003; 108: 925–8. [ Links ]
123. Di Paolo M, Luchini D, Bloise R, Priori SG. Postmortem molecular analysis in victims of sudden unexplained death. Am J Forensic Med Pathol 2004; 25: 182–4. [ Links ]
124. Ackerman MJ, Tester DJ, Porter CJ, Edwards WD. Molecular diagnosis of the inherited long–QT syndrome in a woman who died after near–drowning. N Engl J Med 1999; 341: 1121–5. [ Links ]
125. Chugh SS, Senashova O, Watts A, et al. Postmortem molecular screening in unexplained sudden death. J Am Coll Cardiol 2004; 43: 1625–9. [ Links ]
126. Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovasc Res 2005; 67: 388–96. [ Links ] ]]>