T y dos más fueron heterocigotos para el polimorfismo c.79G>A (p.V27I). En ningún caso se hallaron las mutaciones c.167delT y c.235delC. Conclusiones. Se encontraron cambios de secuencias que correspondieron a dos polimorfismos y a tres mutaciones. La frecuencia de las tres mutaciones investigadas fue menor a lo reportado en la literatura y se encontró una mutación no reportada previamente. Este estudio evidencia la importancia del diagnóstico oportuno con manejo integral, incluyendo el asesoramiento genético con base en estudios moleculares, y resalta la importancia de conocer el perfil genotípico de este grupo de pacientes.]]>
T. Two additional patients had the c.79G>A (p.V27I) polymorphism. Conclusions. Frequency of the three mutations analyzed was lower compared to other populations. Five sequence changes were observed, two polymorphisms and three mutations, one of them novel. This study also demonstrates the relevance of early diagnosis and multidisciplinary management and the importance of determining the genetic basis of this disease in pediatric patients with congenital deafness.]]>
Importancia del diagnóstico de mutaciones en el gen de la conexina 26 en el manejo integral de la sordera congénita no sindrómica
Importance of the diagnosis of connexin 26 gene mutations in the integral management of non-syndromic congenital deafness
Paola Mendelsberg-Fishbein,1 Candy Sue Márquez-Ávila,2 Constanza García-Delgado,1 Adriana Sánchez-Boiso,1 Benjamín Antonio Rodríguez-Espino,3 Edgar Ricardo Vázquez-Martínez,1 Graciela Roque-Lee,2 Silvia Ortiz-Rodríguez,2 María de los Ángeles Fierro-Evans,2 Salvador Castillo-Castillo,2 Rebeca López-Mosqueda,2 Patricia García-Rivera,2 Aura Leonor Flores-Venegas,1* Jesús Aguirre-Hernández,4 Alicia Cervantes-Peredo,5,6 Verónica Fabiola Morán-Barroso1
1 Departamento de Genética
2 Departamento de Audiología y Foniatría ]]>
3 Laboratorio de Investigación en Nefrología
4 Unidad de Investigación en Enfermedades Oncológicas Hospital Infantil de México Federico Gómez
5 Servicio de Genética, Hospital General de México Dr. Eduardo Liceaga
6 Facultad de Medicina, Universidad Nacional Autónoma de México
* Becaria PROBEI
México, D.F., México
Autor de correspondencia: Dra. Verónica Fabiola Morán Barroso
Correo electrónico: vfmoran@himfg.edu.mx, ]]>
veronicafabiolamoranbarroso@yahoo.com.mx
Fecha de recepción: 05-02-13
Fecha de aceptación: 05-03-13
Resumen
Introducción. La sordera congénita es un problema de salud pública. Su incidencia en México es de 2-3 por cada 1000 recién nacidos. El diagnóstico oportuno con el tamiz auditivo neonatal es fundamental para un mejor pronóstico funcional. Aproximadamente 70% de las sorderas congénitas son de origen genético, con herencia autosómica recesiva. La mayoría de estos casos se asocia con mutaciones en el gen GJB2 , que codifica para la proteína conexina 26. Hay tres mutaciones reportadas como las más frecuentes en este gen: c.35delG, c.167delT y c.235delC.
Métodos. Previo consentimiento informado de los pacientes, se obtuvo 1 ml de sangre periférica para la extracción de ADN. Mediante las técnicas de PCR-RFLP o PCR seguida de secuenciación, se buscaron las tres mutaciones más frecuentes del gen GJB2 .
Resultados. Se realizó el estudio molecular en 11 pacientes: Se encontró un cambio en la secuencia codificante en cinco de ellos. Un paciente fue homocigoto para c.35delG; otro resultó heterocigoto para c.35insG, mutación no reportada previamente; un tercero fue heterocigoto para c.34G>T y dos más fueron heterocigotos para el polimorfismo c.79G>A (p.V27I). En ningún caso se hallaron las mutaciones c.167delT y c.235delC.
Conclusiones. Se encontraron cambios de secuencias que correspondieron a dos polimorfismos y a tres mutaciones. La frecuencia de las tres mutaciones investigadas fue menor a lo reportado en la literatura y se encontró una mutación no reportada previamente. Este estudio evidencia la importancia del diagnóstico oportuno con manejo integral, incluyendo el asesoramiento genético con base en estudios moleculares, y resalta la importancia de conocer el perfil genotípico de este grupo de pacientes.
]]> Palabras clave: sordera congénita, conexina 26.
Abstract
Background. Congenital deafness is a public health problem affecting 2-3:1000 newborns in Mexico. Neonatal audiologic screening allows early detection with important implications for the functional prognosis. About 70% of cases of congenital deafness are associated with a genetic etiology with an autosomal recessive pattern of inheritance. Most cases are caused by mutations in the GJB2 gene, which codifies conexin 26. The three most commonly reported mutations in this gene are c.35delG, c.167delT and c.235delC.
Methods. After obtaining informed consent, DNA was extracted from a blood sample, and the three previously mentioned mutations were searched for using PCR-RFLP or PCR followed by sequencing.
Results. Molecular analysis was carried out in 11 patients. In five of these patients, a change in sequence was observed. In none of the patients were c.167delT and c.235delC mutations found. One patient was homozygous for c.35delG and another patient was heterozygous for c.35insG, which is a mutation not previously reported. A third patient was heterozygous for c.34G>T. Two additional patients had the c.79G>A (p.V27I) polymorphism.
Conclusions. Frequency of the three mutations analyzed was lower compared to other populations. Five sequence changes were observed, two polymorphisms and three mutations, one of them novel. This study also demonstrates the relevance of early diagnosis and multidisciplinary management and the importance of determining the genetic basis of this disease in pediatric patients with congenital deafness.
Key words: congenital deafness, connexin 26.
Introducción
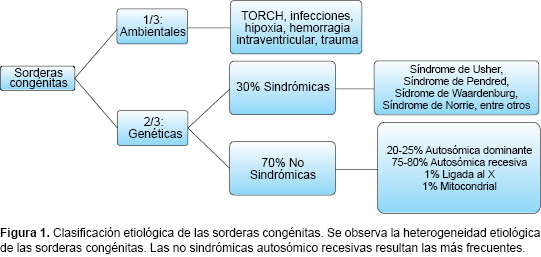
]]> La sordera congénita es un problema de salud pública mundial. Su incidencia varía de acuerdo con los diferentes grupos étnicos.1 Diversos estudios indican que en México alrededor de 2 a 3 de cada 1,000 niños nacen con hipoacusia.2,3 El diagnóstico de la pérdida auditiva durante los primeros meses de vida es de suma importancia ya que, los niños que son identificados a tiempo y a quienes se les da un tratamiento oportuno, tienen un mejor desarrollo de habilidades cognitivas, de lenguaje y sociales. Por esta razón es que en México se implementó el programa Tamiz Auditivo Neonatal e Intervención Temprana 2007-2012 (TANIT), que incluye estrategias para la detección oportuna de la sordera en las Instituciones de la Secretaría de Salud.3 Parte de la complejidad del estudio de las sorderas congénitas es determinar su etiología, la cual impactará en las diversas decisiones de manejo y tratamiento para el paciente, incluyendo la posibilidad del implante coclear y del asesoramiento genético. Diversas guías internacionales establecen que uno de los aspectos fundamentales del manejo integral de los pacientes con sordera es definir el perfil genético, el cual puede variar en cada población (o incluso en cada familia), y tener mutaciones individuales determinadas por las características del grupo étnico y del paciente en particular.4En un tercio de los pacientes con sordera congénita es posible identificar una etiología ambiental, como infecciones, factores adversos perinatales, etcétera. El resto presenta una etiología genética5 (Figura 1), con una gran heterogeneidad. Hasta 70% corresponde a sorderas congénitas no sindrómicas y 30% a sorderas sindrómicas.6 Entre estas, algunas de las principales, por su frecuencia o por su alto impacto clínico, son los síndromes de Waardenburg (OMIM 193500), de Usher (OMIM 276900), de Pendred (OMIM 274600), banquiootorenal (OMIM 113650), de Jervell y Lange-Nielsen (OMIM 220400), de Alport (203780) y las mitocondriopatías.
Las sorderas congénitas no sindrómicas (aquellas en las que la sordera es la única manifestación) tienen una gran heterogeneidad genética y se han identificado más de 100 loci relacionados con su etiología. El patrón de herencia mayormente identificado, presente en más de 70% de los casos, es el autosómico recesivo.7 Para la designación de los casos de sordera congénita y para resaltar su etiología, la nomenclatura utilizada internacionalmente usa las siglas DFN, por deafness (sordera, en inglés), seguida por una letra. Así, la letra A se utiliza para definir el patrón de herencia autosómico dominante (DFNA), la B para autosómico recesivo (DFNB) y la X para ligado al cromosoma X (DFNX). Finalmente, el número siguiente con que se designa cada uno corresponde al orden en el que se mapeó el gen responsable. Adicionalmente, la sordera puede deberse a una mutación en el genoma mitocondrial, por lo que puede considerarse como una mitocondropatía.
Diferentes estudios en Europa, Asia, Estados Unidos y Sudamérica demuestran que de 50%-80% de los casos de sordera congénita no sindrómica son autosómicos recesivos y se dan por mutaciones en el locus DFNB1 (OMIM: 220290) en 13q11-q12.8 En esta región se encuentran dos genes: GJB2 , que codifica para la proteína conexina 26 y se encuentra mutado en 98% de los casos de DFNB1 y el gen GJB6 , que codifica la proteína conexina 30 y causa el 2% restante de las sorderas DFNB1. Se ha observado que hay otras mutaciones en este mismo gen que producen casos de sordera autosómica dominante (DFNA3)9 y otras más que causan sorderas sindrómicas que se acompañan de alteraciones dermatológicas.10
]]> Se han identificado tres mutaciones autosómicas recesivas en GJB2 como las más frecuentes: c.35delG en población caucásica, c.167delT en judíos Ashkenazi y c.235delC en poblaciones asiáticas.6-12 Estas tres mutaciones producen un corrimiento en el marco de lectura y un codón de alto prematuro. La mutación más frecuentemente reportada es la c.35delG, que podría corresponder a hasta 70% de los alelos mutantes del gen GJB2 relacionados con sordera.13A la fecha no hay estudios publicados sobre pacientes mexicanos con sordera congénita. Si bien, existen algunos trabajos preliminares que se han presentado en congresos nacionales,14 tampoco se conoce hasta el momento cuál es la mutación más frecuente en nuestra población. Por ello el ''Grupo de Estudios Multidisciplinario de Sordera Congénita'' del Hospital Infantil de México Federico Gómez (HIMFG) planteó el protocolo de investigación para determinar, en pacientes con sordera congénita no sindrómica atendidos en el hospital, las tres mutaciones más frecuentes en GJB2 .
Métodos
Posterior a la autorización institucional por los Comités de Investigación, Ética y Bioseguridad, del protocolo de investigación correspondiente (HIM/2010/011), se identificaron los pacientes que acudieron en los últimos 5 años (2007 a 2012) a la institución con diagnóstico de sordera congénita no sindrómica, sin factores de riesgo perinatales aparentes para la misma. Previo consentimiento informado, se tomó muestra de 1 ml de sangre periférica y se extrajo el ADN, utilizando el kit de Puregene (Qiagen®) de acuerdo con las especificaciones del fabricante. Posteriormente, se diseñaron los oligonucleótidos para realizar la amplificación por técnica de PCR de los tres fragmentos del gen GJB2 en donde se encuentran las tres mutaciones más frecuentemente reportadas (Cuadro 1). Para la identificación de la mutación c.35delG se realizó la secuenciación (en ambas cadenas) de un fragmento de 210 pb de la región codificante. La identificación de las mutaciones c.235delC y c.167delT se realizó mediante la técnica de análisis de los polimorfismos en la longitud de los fragmentos de restricción (RFLP). Las enzimas utilizadas fueron:
1) Pst l para la identificación de la mutación c.167delT, que reconoce la secuencia 5´CTGCAG3´ y corta la secuencia normal, produciendo un fragmento de 116 pb y otro de 47 pb. En el producto de PCR con la mutación c.167delT no se produce el corte.
2) Apa I se utilizó para la búsqueda de la mutación c.235delC. Su secuencia de reconocimiento es 5´GGGCCC3´. En la secuencia normal produce fragmentos de 151 y 59 pb. Si se encuentra la mutación c.235delC, la enzima no realiza el corte.
Los RFLP fueron visualizados en gel de agarosa al 2% teñido con bromuro de etidio, utilizando un marcador de peso molecular de 50 pares de bases como referencia para confirmar el tamaño esperado de los fragmentos generados.
Resultados
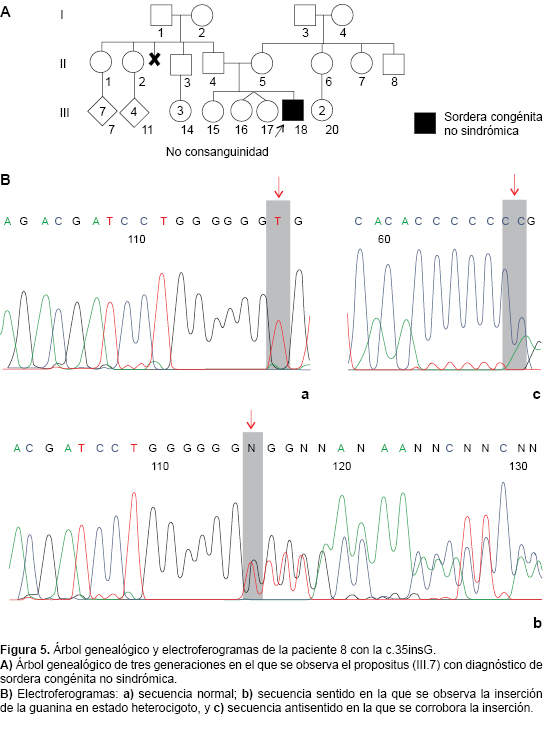
]]> Se identificaron 96 pacientes con diagnóstico de sordera probable no sindrómica. En 63 de ellos se identificó una causa ambiental o una entidad sindrómica, por lo que se excluyeron del estudio. De los 33 casos restantes de sordera congénita no sindrómica (Cuadro 2), se efectuó el análisis molecular en once de ellos (Figura 2). En todos los casos, el estudio por PCR-RFLP permitió descartar la presencia de las mutaciones c.167delT y c.235delC. En cinco casos se identificaron cambios en la secuencia de nucleótidos del fragmento amplificado de 210 pb. Tres de las variantes correspondieron a mutaciones y dos a polimorfismos. En el paciente 5 se identificó la mutación c.35delG en estado homocigoto (Figura 3). En el paciente 4 se encontró la mutación c.34G>T en estado heterocigoto (Figura 4). En el paciente 8, la mutación c.35insG en estado heterocigoto (Figura 5). Esta última no ha sido reportada previamente en la literatura. En los pacientes 2 y 3 se encontró el polimorfismo c.79G>A en estado heterocigoto (Cuadro 2).


Discusión
Del grupo de pacientes con sordera del HIMFG, se identificaron 96 pacientes con diagnóstico probable de sordera congénita no sindrómica. De ellos, sólo 33 casos fueron candidatos a estudio molecular, por considerarse de etiología exclusivamente genética. Es interesante señalar que del grupo original de 96 candidatos, en 63 fue posible identificar una causa, factor de riesgo ambiental o entidad sindrómica, por lo que fueron descartados como candidatos para el estudio molecular del gen de conexina 26. Sin embargo, esta elevada frecuencia de causas no genéticas de sordera es un dato importante del perfil de la población que atiende nuestro hospital y que difiere de lo reportado a nivel mundial, donde la mayor parte de los casos corresponde a sordera congénita no sindrómica o no asociada a factores predisponentes perinatales. Esta situación podría explicarse por el tipo de institución, dado que los pacientes atendidos en el HIMFG son referidos para su evaluación de todo el país, particularmente los casos que requieren tercer nivel de atención por su complejidad o características asociadas, lo cual indica una diferencia importante en el tipo de población estudiada. La mayor parte de los pacientes en los que se realizaron estudios a nivel molecular provinieron del Estado de México (6 pacientes) y Michoacán (3 pacientes). Esto se explica, en el primer caso, por la cercanía geográfica con la institución y, en el segundo, por el convenio de manejo y tratamiento de estos pacientes que la institución tiene con ese estado, siendo también esto un posible sesgo poblacional.
De los once pacientes en los cuales se realizó el estudio molecular, únicamente en tres se estableció el diagnóstico por medio de tamizaje auditivo neonatal; el resto se diagnosticó de forma más tardía (rango de edad entre 2 y 5 años). De este último grupo, 6 no han logrado desarrollar lenguaje hablado. Comparativamente, algunos de los pacientes diagnosticados tempranamente, que utilizaron auxiliares auditivos e ingresaron al programa de evaluación de implante coclear, y empezaron su rehabilitación y tratamiento oportuno desde el momento de la sospecha clínica, ya lo han desarrollado. Esto resalta la enorme importancia de la detección temprana de la hipoacusia mediante el programa de tamizaje auditivo neonatal, lo que confiere un mejor pronóstico para la integración y calidad de vida de los pacientes.
]]> En tres casos se corroboró la existencia de consanguinidad entre los padres de los pacientes y en cuatro pacientes hay antecedentes heredofamiliares de sordera con afectados en la misma generación, lo que sugiere un patrón de herencia autosómico recesivo.En la gran mayoría de nuestros pacientes la sordera fue de tipo sensorineural profunda bilateral. Este es el tipo de sordera más frecuente en los casos de sordera congénita no sindrómica, por lo que la población analizada refleja lo reportado previamente en la literatura.4
Se identificaron mutaciones en el gen GJB2 en tres de los once pacientes analizados, lo cual corresponde a 27% de los casos. Esto concuerda con una proporción similar a la esperada, por lo reportado en la literatura, ya que se calcula que hasta 20% de los casos de sordera congénita no sindrómica se originan por mutaciones en este gen.15 Sin embargo, la muestra analizada para este estudio es muy pequeña para poder establecer frecuencias, además de que sólo se buscaron las tres mutaciones más frecuentes, incluyendo la secuenciación de un fragmento de 210 pb, por lo que no se puede descartar la presencia de mutaciones en las regiones no analizadas del gen.
En este sentido, sólo en uno de nuestros casos se identificó la mutación c.35delG, la más frecuentemente asociada con la sordera no sindrómica autosómica recesiva. Esto corresponde al 9% de los casos estudiados, lo que constituye un porcentaje menor al reportado en la literatura, que corresponde de 28 a 63% de las mutaciones en GJB2 , dependiendo de la población.6
Hasta donde se conoce, no existen publicaciones que reporten el tipo o frecuencia de mutaciones asociadas con casos de sordera congénita en población mexicana. De manera reciente, dos grupos de investigación en México presentaron en el Congreso Nacional de Genética Humana 201214-16 sus resultados, en los que solamente 3% de los casos fueron homocigotos para la c.35delG. Esto corresponde a un porcentaje todavía menor al que se encontró en este estudio. Sin embargo, como ya se mencionó, nuestra muestra es aún muy pequeña para determinar las frecuencias. En nuestro caso, llama la atención que la mutación se encontró en estado homocigoto y que los padres de la paciente no son consanguíneos, ni provienen del mismo estado de la República, por lo que sería importante determinar la frecuencia de portadores (heterocigotos) sanos de la mutación en la población mexicana. En uno de los pacientes se encontró el cambio c.34G>T en estado heterocigoto. Este ha sido previamente reportado,17 pero no se ha demostrado su efecto patológico. Actualmente está clasificado como un cambio probablemente patológico (rs104894408). Esta mutación de sentido equivocado, causa un cambio de glicina por cisteína en la posición 12 dentro de la cadena de aminoácidos de la proteína (p.G12C). La glicina es un aminoácido que tiene cadenas laterales alifáticas, es hidrofílico y polar; mientras que la cisteína es también un aminoácido hidrofílico y polar, pero tiene cadenas laterales con átomos de azufre, lo que le confiere la capacidad de producir enlaces disulfuro, por lo que este cambio puede generar alteraciones en la estructura proteica. Este cambio fue reportado en pacientes con sordera congénita no sindrómica, en un estudio realizado por Putcha y colaboradores.17 En él, analizaron los genes GJB2 y GJB6 en más de siete mil pacientes con sordera congénita no sindrómica en Estados Unidos. El cambio p.G12C correspondió, como única alteración, a solamente 0.4% de todas la mutaciones detectadas en los dos genes estudiados.
Se encontró otra mutación que corresponde a una inserción de una guanina dentro de la secuencia de las seis guaninas en las que ocurre la c.35delG. Esta mutación no ha sido reportada y causa un cambio en el marco de lectura con un codón de alto prematuro en el codón 67. En este caso, la inserción se encontró en estado heterocigoto. Ante esta alteración existen varios aspectos a considerar. No se puede descartar que se trate de un heterocigoto compuesto, ya que no se realizó el análisis molecular de todo el gen (únicamente de dos mutaciones específicas por PCR-RFLP y de un fragmento por secuenciación). Tampoco puede descartarse la presencia de otra mutación en la otra copia del gen, mutación que estaría en una región distinta de las estudiadas.6
Otra posibilidad es que se trate de un doble heterocigoto ya que las conexinas funcionan en grupos de 6 proteínas y se pueden unir a otras conexinas, particularmente la conexina 30, formando un conexón heteromérico.18 Se han descrito casos de dobles heterocigotos con una mutación en un alelo del gen GJB2 y otra mutación en un alelo de otro gen de esta familia, principalmente en el gen GJB6 , que codifica para la conexina 30 y que se encuentra en el mismo locus. 19
Finalmente, se identificó en dos pacientes el polimorfismo c.79G>A en estado heterocigoto. Es interesante que este polimorfismo se ha reportado en el 41% de los pacientes,14 por lo que es probable que este cambio sea frecuente en la población mexicana y no esté asociado a alguna patología. Sin embargo, se ha planteado la posibilidad de que la presencia de este SNP con otro cambio en la secuencia del gen GJB2 , podría causar sordera,20 por lo que será importante determinar la frecuencia de este cambio en controles mexicanos.
En conclusión, este estudio preliminar representa el primer reporte de frecuencia de las mutaciones c.35delG, c.167delT y c.235delC del gen GJB2 en pacientes mexicanos con sordera congénita. Se descartó en este grupo la presencia de las mutaciones c.167delT y c.235delC y en 5 pacientes (45% de los casos) se encontraron cambios en su secuencia: dos polimorfismos y tres mutaciones, una de ellas no reportada previamente. Esto resalta la importancia de conocer el perfil genotípico de nuestra población.
Agradecimientos
]]> El proyecto agradece a la Dirección de Investigación por el apoyo con una beca del Programa de Becas de Inicio a la Investigación (PROBEI) para la alumna Aura Leonor Flores Venegas. Este proyecto contó con el apoyo de fondos federales.
REFERENCIAS
1. García-Pedroza F, Peñaloza-López Y, Poblano A. Los trastornos auditivos como problema de salud pública en México. An Orl Mex 2003;48:20-29. [ Links ]
2. Poblano A, Arteaga C, García-Sánchez G. Prevalence of early neurodevelopmental disabilities in Mexico: a systematic review. Arq Neurol Psiquiatr 2009;67:736-740. [ Links ]
3. Consejo Nacional para el Desarrollo y la Inclusión de las Personas con Discapacidad (CONADIS). Disponible en: www.conadis.salud.gob.mx [ Links ]
4. American College of Medical Genetics. Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss. Genetic Evaluation of Congenital Hearing Loss Expert Panel. ACMG statement. Genet Med 2002;4:162-171. [ Links ]
]]>5. Raviv D, Dror AA, Avraham KB. Hearing loss: a common disorder caused by many rare alleles. Ann NY Acad Sci 2010;1214:168-179. [ Links ]
6. Mahdieh N, Rabbani B. Statistical study of 35delG mutation of GJB2 gene: a meta-analysis of carrier frequency. Int J Audiol 2009;48:363-370. [ Links ]
7. Cordeiro-Silva Mde F, Barbosa A, Santiago M, Provetti M, Rabbi-Bortolini E. Prevalence of 35delG/GJB2 and del (GJB6-D13S1830) mutations in patients with non-syndromic deafness from a population of Espírito Santo-Brazil. Braz J Otorhinolaryngol 2010;76:428-432. [ Links ]
8. Van Laer L, Coucke P, Mueller RF, Caethoven G, Flothmann K, Prasad SD, et al. A common founder for the 35delG GJB2 gene mutation in connexin 26 hearing impairment. J Med Genet 2001;38:515-518. [ Links ]
9. Welch K.O., Marin R.S., Pandya A., Arnos K.S. Compound heterozygosity for dominant and recessive GJB2 mutations: effect on phenotype and review of the literature. Am J Med Genet A 2007;143A:1567-1573. [ Links ]
]]>10. Kenna MA, Rehm HL, Robson CD, Frangulov A, McCallum J, Yaeger D, et al. Additional clinical manifestations in children with sensorineural hearing loss and biallelic GJB2 mutations: who should be offered GJB2 testing? Am J Med Genet A 2007;143A:1560-1566. [ Links ]
11. Tsukada K, Nishio S, Usami S; Deafness Gene Study Consortium. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet 2010;78:464-470. [ Links ]
12. Ensembl Genome Browser. Disponible en: www.ensembl.org [ Links ]
13. Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet 1997;6:2173-2177. [ Links ]
14. Arenas-Sordo M, Hernández-Zamora E, Gutiérrez-Tinajero D, Murphy-Ruiz P, Leyva-Juárez X, Huesca-Hernández F, et al. Mutaciones de los genes GJB2 y GJB6 en pacientes con hipoacusia bilateral prelinguística del INR. Memorias del III Congreso Internacional de Investigación en Rehabilitación, Ciudad de México. Noviembre, 2012. Disponible en: http://www.inr.gob.mx/Descargas/ciir/memorias3erCongreso.pdf [ Links ]
15. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 1998;62:792-799. [ Links ]
]]>16. Martínez-Saucedo M, González-Huerta LM, Berruecos P, Rivera-Vega MR, Cuevas-Covarrubias SA. Análisis de la deleción del gen GJB6 en pacientes heterocigotos para el gen GJB2 con hipoacusia neurosensorial no sindrómica en una muestra de población mexicana. XXXVII Congreso Nacional de Genética Humana, Guadalajara, Jalisco. Noviembre, 2012. [ Links ]
17. Putcha GV, Bejjani BA, Bleoo S, Booker JK, Carey JC, Carson N, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med 2007;9:413-426. [ Links ]
18. Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol 2009;1:a002576. [ Links ]
19. Stevenson VA, Ito M, Milunsky JM. Connexin-30 deletion analysis in connexin-26 heterozygotes. Genet Test 2003;7:151-154. [ Links ]
20. Dahl HH, Tobin SE, Poulakis Z, Rickards FW, Xu X, Gillam L, et al. The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children. J Med Genet 2006;43:850-855. [ Links ]
]]>