Porfiria variegata en Chile: identificación de mutaciones en el gen protoporfirinógeno oxidasa y su implicancia diagnóstica
Identification of mutations in the protoporphyrin oxidase gene and its diagnostic implications in porphyria variegata in Chile
Carlos Wolff,* Jorge Frank,**,*** Pamela Poblete–Gutiérrez**
* Departamento de Medicina Occidente, Universidad de Chile, Hospital San Juan de Dios. Santiago, Chile.
** Departamento de Dermatología, Academisch Ziekenhuis Maastricht, P. Holanda.
]]> *** Centro de Porfirias, Universitatsklinikum der RWTH, Aachen, Alemania
Reimpresos:
Dr. Carlos Wolff
Departamento de Medicina Occidente Universidad de Chile
Casilla 33052, Santiago, Chile
Correo electrónico: cwfl255@yahoo.es
Recibido el 8 de diciembre de 2005.
Aceptado el 22 de mayo de 2006.
ABSTRACT
Variegate porphyria (VP) results from a hereditary deficiency of protoporphyrinogen oxidase (PPOX) that is transmitted in an autosomal dominan fashion. The diagnosis is based on the clinical symptoms and is confirmed biochemically. Sometimes, however, these diagnostic tools reveal limitations in establishing the definitive diagnosis of the prevailing type of acute porphyria. In these patients, molecular genetic analyses can be useful. We performed molecular genetic studies in 13 Chilean families by PCR amplification of the PPOX gene, conformation sensitive gel electrophoresis, and automated DNA sequencing. In five symptomatic patients from different families, respectively, the biochemical data confirmed the diagnosis of VP. In seven other families, however, the biochemical studies were not conclusive. Furthermore, the original biochemical analysis in one clinically severely affected patient from a further family even suggested the diagnosis of erythropoietic protoporphyria (EPP). Beside the respective index patients, we studied 78 asymptomatic family members and 50 healthy, unrelated individuals for control purposes. In five families, the previous diagnosis of VP could be confirmed genetically. Further, half of the asymptomatic relatives revealed a mutation in the PPOX gene, consisting of three missense mutations and two deletion mutations. Mutation R168H that had been already described previously in German VP families was found in a Chilean family of German origin. Further, two novel missense mutations, designated L74P and G232S, could be detected. In four Chilean families, we found the deletion 1330deICT that had also been previously described in three Swedish VP families. The second deletion, 1239delTACAC, has not been described anywhere else but Chile and could be identified in seven families. One patient who was initially diagnosed with EPP turned out to be a compound heterozygote for mutations on both alíeles of the PPOX gene. In conclusion, our molecular genetic analyses unequivocally confirmed the diagnosis of VP in seven families who originally had revealed inconclusive biochemical data. Further, early genetic analysis allows for the identification of asymptomatic mutation carriers, thereby offering the possibility of adequate counselling and the prevention of potentially life–threatening acute porphyric attacks.
Key words. Variegate porphyria. Molecular genetic diagnosis.
RESUMEN
La porfiria variegata (PV), enfermedad de origen genético con forma de herencia autosómica dominante, se debe a deficiencia en la actividad protoporfirinógeno oxidasa (PPOX). Su diagnóstico se basa en antecedentes clínicos y se confirma con análisis bioquímicos. Éstos, en algunos casos, pueden presentar limitaciones para establecer el diagnóstico definitivo de la variedad de porfiria aguda, situación en que el estudio genético molecular puede resultar útil. Se efectuó estudio genético en trece familias chilenas usando amplificación del gen PPOX por PCR, electroforesis conformacional y secuenciación automática de DNA. Cinco de estas familias incluían pacientes índices sintomáticos con diagnóstico bioquímico establecido de PV; otras siete familias incluían pacientes índices con estudio bioquímico no concluyente de la variedad de porfiria aguda y, finalmente, una familia con diagnóstico previo de protoporfiria eritropoyética (PPE). Además, se estudiaron 78 familiares asintomáticos y 50 personas sanas, no relacionadas, como controles. En cinco familias el estudio genético confirmó el diagnóstico bioquímico previo de PV. El 50% de los familiares asintomáticos resultaron ser portadores de una mutación en el gen PPOX. Se identificaron tres mutaciones por sustitución de bases: la R168H, descrita en familias de origen alemán y dos nuevas mutaciones, designadas L74P y G232S. También se identificaron dos mutaciones por deleción de bases designadas 1330delCT y la 1239delTACAC. La primera, que había sido descrita previamente en tres familias suecas, se encontró en cuatro familias chilenas. La segunda se encontró en siete familias y no ha sido descrita previamente. El estudio genético permitió mostrar que un paciente que originalmente fue diagnosticado con PPE correspondía a un heterocigoto compuesto para dos mutaciones en el gen PPOX. En conclusión, los estudios moleculares permitieron confirmar el diagnóstico de PV en cinco familias, efectuar diagnóstico de PV en familias en las cuales los datos bioquímicos no eran concluyentes, corregir el diagnóstico original en una familia e identificar portadores asintomáticos entre los familiares de los pacientes índices. Los estudios genéticos moleculares ayudan a realizar un adecuado consejo genético a pacientes y familiares y hace posible practicar prevención de las crisis agudas de porfiria, las que son potencialmente mortales.
Palabras clave. Porfiria Variegata. Diagnóstico genético molecular.
INTRODUCCIÓN
]]> Las porfirias constituyen un grupo heterogéneo de enfermedades de origen genético o adquirido, debidas a disminución de la actividad de una de las enzimas de la vía de síntesis del grupo heme (Figura 1), lo que lleva a la acumulación intra y extracelular de metabolitos intermediarios, los que presentan acción citotóxica y fotosensibilizante.De acuerdo con la expresión clínica, las porfirias se clasifican en agudas y no agudas (Cuadro 1). Las primeras se caracterizan por presentar crisis agudas, con intenso dolor abdominal de tipo cólico, náuseas, vómitos, hiponatremia, hipertensión arterial y variado compromiso neurológico, incluido el coma. Estos ataques son comúnmente gatillados, entre otros factores, por la ingesta de medicamentos, especialmente los anestésicos. En las porfirias no agudas los síntomas más característicos son las alteraciones cutáneas, especialmente fotosensibilidad, labilidad cutánea, hipertricosis e hiperpigmentación.
Habitualmente el diagnóstico se basa en los antecedentes clínicos y se confirma con la medición de los distintos metabolitos intermediarios de las porfirinas en orina, heces y sangre y, en ocasiones, con determinaciones de la actividad de enzimas involucradas.1 El análisis genético molecular se ha incorporado recientemente, contribuyendo a mejorar el diagnóstico.2
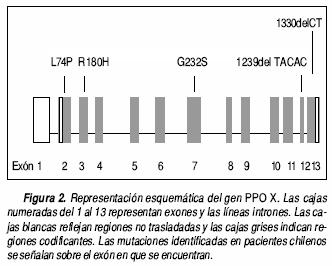
Entre las distintas variedades de porfirias agudas, la variegata (PV) (OMIM 176200) es la que con mayor frecuencia hemos diagnosticado en Chile,3 con un total de 24 familias, 30 casos sintomáticos y 48 portadores asintomáticos de la enfermedad. Los pacientes con PV presentan crisis porfíricas y además lesiones cutáneas en áreas fotoexpuestas, producto del aumento de la circulación sanguínea y acumulación de porfirinas en la piel que resulta en síntomas cutáneos incluyendo ampollas, erosiones, escoriaciones, milias, cicatrices e hiperpigmentación. Estas alteraciones no son exclusivas de la PV, pudiendo estar también presente en la porfiria cutánea tarda y en la coproporfiria hereditaria. La PV presenta un modo de herencia autosómica dominante con expresión incompleta, ya que no todas las personas que heredan el defecto genético expresan síntomas clínicos. La enfermedad se debe a la disminución de la actividad de la penúltima enzima en la vía de síntesis del heme, la protoporfirinógeno oxidasa (PPOX) (EC 1.3.3.4),4 debida a mutaciones heterocigotas en el gen PPOX. La PPOX se encuentra en eucariontes a nivel de la membrana interna mitocondrial y cataliza la conversión del protoporfirinógeno IX a protoporfirina IX. El gen PPOX se ubica en el cromosoma Iq22 y fue clonado en 1995, consta de 13 exones y 12 intrones2 (Figura 2).
Normalmente la PV se expresa después de la pubertad,3 aunque se han descrito algunos casos excepcionales en niños.5 Pacientes heterocigotos presentan una disminución de alrededor de 50% de la actividad normal de la enzima, mientras que pacientes heterocigotos compuestos u homocigotos, es decir, portadores de mutaciones en ambos alelos, presentan un intenso déficit enzimático y una expresión fenotípica más intensa, expresada desde la infancia y en ocasiones con déficit de inteligencia y alteraciones óseas.6–8
Se ha reportado un gran número de mutaciones en el gen PPOX, siendo la mayoría de tipo privado, propias de un individuo o una familia. Sólo en Sudáfrica y recientemente en Chile, con ayuda de análisis de haplotipo, se ha logrado identificar mutaciones fundadoras, es decir, que derivan de un ancestro común.9,10
El diagnóstico de la PV presenta en algunos casos gran dificultad, especialmente durante las crisis agudas, ya que con sólo los resultados bioquímicos no es posible distinguirla con claridad de otras formas agudas de porfiria como son la coproporfiria hereditaria y la aguda intermitente. Además, muchos pacientes tienden a normalizar sus parámetros bioquímicos entre una crisis y otra, situación que también dificulta el correcto diagnóstico.11,12
El objetivo de nuestro trabajo fue caracterizar 13 familias chilenas con diagnóstico de porfiria desde un punto de vista genético molecular, consistente en la búsqueda de mutaciones en el gen PPOX y, además, analizar el aporte a la clínica de la realización de estos estudios.
]]>PACIENTES Y MÉTODOS
Selección de casos
Se estudiaron 13 familias chilenas, no relacionadas entre sí, con diagnóstico de porfiria. En cinco de ellas, en los casos índices se estableció el diagnóstico de PV por los antecedentes clínicos que incluían crisis aguda y un perfil característico de los exámenes de laboratorio. En siete familias los casos índices presentaron crisis aguda e intensas alteraciones en la excreción de porfirinas, pero por haber sido efectuados durante las crisis, no permitieron establecer un patrón típico de una variedad de porfiria específica, catalogándoselas como "porfirias agudas". Se estudió una 13a familia cuyo paciente índice presentó, desde niño, alteraciones cutáneas expresadas en intensa fotosensibilidad y exámenes bioquímicos con un patrón propio de PPE, pero en quien no se encontró mutaciones en el gen de la ferroquetalasa (FC), que habría sido la característica de genética propia de la PPE. Además, se estudiaron 78 familiares de primer grado de los casos índices, asintomáticos sin alteraciones de laboratorio y 50 personas sanas no relacionadas entre sí con los casos índices como controles. El criterio para incluir una familia en el estudio fue la presencia de al menos un miembro con sintomatología compatible con alguna variedad de porfiria aguda y con significativa excreción de porfirinas y precursores, esto es, elevación urinaria de porfirinas totales, ácido delta aminolevulínico (ALA) y porfobilinógeno (PBG) en episodios de crisis y una significativa elevación de las protoporfirinas en relación con las coproporfirinas en heces en los periodos asintomáticos, aunque ésta no fuera concluyente para establecer la variedad de porfiria.
Análisis bioquímico
Las determinaciones de porfirinas y precursores en muestras de orina, sangre y heces se realizó por procedimientos descritos anteriormente.3
Material y extracción del DNA
De cada paciente, familiar y control se obtuvo, previo consentimiento informado, 5 mL de sangre total en tubos con EDTA, procediéndose a extraer DNA genómico utilizando una técnica estandarizada.13
]]>Amplificación del DNA
La amplificación de los exones codificantes y zonas inmediatas adyacentes de los intrones del gen PPOX se realizó utilizando 12 pares de partidores diseñados y publicados previamente.10
Todas las amplificaciones se llevaron a cabo en un termociclador Biometra TGradient (Whatman Biometra, Góttingen, Alemania) de acuerdo con el siguiente programa: predenaturación a 95 °C por 5 minutos, seguido de 35 ciclos de denaturación a 95 °C por 45 segundos, hibridación de los cebadores a temperaturas específicas por 45 segundos y extensión a 72 °C por un minuto. La extensión final se realizó a 72 °C por siete minutos. Cada reacción de amplificación se realizó con aproximadamente 100 ng de DNA genómico, 100 ng/mL de cada partidor y 45 mL de Platinum Taq PCR Super Mix, que contenía 40 mM Tris pH 8.4, 100 mM KC1, 1.6 mM dNTPs, 6 mM MgCl2 y 50 U/mL Platinum Taq polymerase (Invitrogen Life Technologies, Karlsruhe, Alemania) en un volumen final de 50 mL.
Detección de mutaciones
Los productos PCR fueron sometidos a análisis conformacional en un gel de electroforesis (CSGE) descrito previamente.14 Los productos PCR, que mostraron una formación heterodúplex en el análisis de CSGE, fueron purificados en una primera etapa, utilizando un High Pure PCR product purification kit (Roche, Basel, Suiza) y, posteriormente, sometidos a una hiperpurificación en columnas Edge Centriflex (Edge BioSystems, Gaithersburg, MD, USA). Los productos fueron finalmente secuenciados utilizando polímero POP–6 en un secuenciador automático Genetic Analyzer ABI Prism 310 (Applied Biosystems, Foster City, CA). Para confirmar las dos deleciones, 1239delTACAC y 1330delCT y excluir la posibilidad de que las alteraciones encontradas corresponden a polimorfismos del gen PPOX, se realizó análisis CSGE y secuenciación automática de los exones correspondientes en muestras de DNA de 50 personas controles. Adicionalmente, se efectuó digestión con endonucleasas de restricción (BstNI, Ncol y Mnll) para confirmar la causalidad de las mutaciones L74P, R168H y G232S (Cuadro 2).
RESULTADOS
Las mutaciones encontradas en este estudio consisten de dos deleciones, 1330delCT y 1239delTA–CAC y tres mutaciones puntuales por sustitución de bases, L74P, R168H y G232S, que conducen estas últimas a las siguientes sustituciones de aminoácidos en la proteína PPOX: lisina en posición 74 por prolina, arginina en posición 168 por histidina y glicina en posición 232 por serina, respectivamente. Un resumen de las mutaciones se señala en el cuadro 2 y su distribución en el gen PPOX en la figura 2.
]]> Once (82%) de los 13 casos índices de las familias con PV son mujeres.El estudio genético molecular confirmó el diagnóstico de PV en las cinco familias con pacientes índices diagnosticados previamente por los antecedentes bioquímicos y clínicos y, además, en las siete familias caracterizadas previamente como agudas sin definir la variedad. En el paciente diagnosticado al nacer como PPE, el estudio genético identificó dos mutaciones en el gen PPOX, la deleción 1330delCT en el alelo materno y una sustitución en el exón 7 en el alelo paterno consistente en la transición del nucleótido 694 G a A en el cDNA del PPOX, lo que conduce a la mutación descrita G232S.15En 36 (46%) familiares de los 78 estudiados que pertenecían a familias con PV, el estudio genético molecular identificó alguna mutación en el gen PPOX, indicando que se trata de portadores asintomáticos.
DISCUSIÓN
A diferencia de lo que se ha descrito en otros países, en Chile, así como en la población caucásica de Sudáfrica, la PV es la variedad más frecuente entre las porfirias de tipo agudo.3
En este estudio se identificaron cinco mutaciones en el gen PPOX. Tres de ellas por sustitución de bases: la mutación R168H encontrada en una familia chilena de origen alemán que ha sido descrita previamente por otros autores en familias de origen alemán, holandés y norteamericano.16 Por el contrario, las mutaciones L74P y G232S no se han descrito anteriormente. La mutación L74P que conduce a un cambio de una leucina por una prolina se encuentra entre una serie de tres leucinas en posición 72–74 de la proteína PPOX. Una mutación similar ya fue reportada en posición 73, designada L73P.17 Es interesante haber identificado estas dos alteraciones genéticas porque los aminoácidos leucina en posición 74 y glicina en posición 232 de la proteína de la PPOX se encuentran conservadas en la evolución en varias especies, incluyendo el Homo sapiens y el ratón,18 además, mutaciones similares y comparables ya se han descrito en el gen PPOX. La importancia de la glicina en posición 232 para la mantención de la actividad de la PPOX es apoyada por el hecho que una mutación en el mismo aminoácido, designado G232R, consistente en la sustitución de la glicina 232 por una arginina, ha sido informado en un paciente francés con PV.19 Estos hallazgos evidencian la importancia de los aminoácidos en posición 74 y 232 de la proteína PPOX en la mantención de la función fisiológica.
Las deleciones identificadas corresponden a mutaciones recurrentes, ya que la mutación 1239del–TACAC se encontró en siete familias chilenas con PV y la 1330delCT en cuatro familias. Con base en estudios de haplotipo, la deleción 1239delTACAC la hemos descrito previamente como una mutación de tipo fundadora, lo que sugeriría que las familias que la portan tienen, probablemente, un ancestro común.10 La deleción 1330delCT, descrita previamente en tres familias suecas,20 por haber sido encontrada en cuatro familias chilenas, es posible que también corresponda a una mutación fundadora, lo que estamos tratando de confirmar con un estudio de haplotipo. La identificación de al menos estas dos mutaciones recurrentes en la población chilena podría dar cuenta por qué la PV es la variedad más frecuente de las porfirias de tipo aguda en Chile.
En este trabajo, el estudio genético molecular permitió identificar la variedad de porfiria en siete pacientes en que los antecedentes clínicos y el estudio bioquímico fueron insuficientes para efectuar el diagnóstico de la variedad de porfiria de tipo aguda. El hecho de haber encontrado sólo un portador de la mutación L74P sintomático (caso índice) y otros siete asintomáticos en los 14 miembros de la familia No. 2 (Cuadro 2), reafirma la idea que las alteraciones genéticas en la PV son de baja penetrancia.21
En forma similar a lo que describe la literatura, en esta serie se encontró que 82% de los pacientes índices son mujeres. Aunque la forma de herencia de la PV no está ligada al sexo, la mayor frecuencia de casos índices de sexo femenino, como lo encontrado en la serie estudiada en este trabajo, se ha atribuido a alteraciones endocrinas, fisiológicas o patológicas, que inducen la generación de crisis agudas en las porfirias agudas.22
El estudio genético molecular permitió corregir el diagnóstico realizado por los antecedentes clínicos y los exámenes de laboratorio en el caso del niño inicialmente diagnosticado como portador de PPE. Esta duda diagnóstica se entiende porque la expresión fenotípica era distinta a la que corrientemente se encuentra en la PV y el estudio genético demostró que se trataba de un heterocigoto compuesto, esto es la existencia simultáneamente de dos mutaciones distintas en ambos alelos del gen PPOX. La identificación de la verdadera variedad de porfiria que presenta este niño, gracias al estudio genético molecular, es de particular importancia porque la evolución de la enfermedad y los riesgos clínicos son diferentes entre el diagnóstico original de PPE y el de heterocigoto compuesto para PV.15
]]> Es interesante destacar que 50% de los familiares asintomáticos de pacientes índices con PV se identificó como portadores de mutaciones en el gen PPOX, lo que los hace susceptibles, eventualmente, de tener una crisis aguda si se ven expuestos a factores desencadenantes de estas crisis.23 Por esto, el principal beneficio clínico de los estudios genéticos moleculares consiste en poder identificar a estas personas portadoras asintomáticas, por cuanto ello permite prevenir las crisis agudas, las que tienen riesgo de muerte, evitando los factores desencadenantes. El manejo de la información obtenida en estas personas que son portadoras de una alteración genética, pero que no presentan la enfermedad, incluso es posible que nunca la tengan, debe manejarse con especial cuidado en relación con su confidencialidad. También se debe estar preparado para efectuar un adecuado consejo genético, tanto a los pacientes como a los familiares portadores de mutaciones, cuando ellos lo soliciten.1Es recomendable que los portadores de porfirias agudas sean advertidos de su condición, clarificarles su significado clínico, esto es, que la enfermedad puede no expresarse clínicamente si evitan los factores desencadenantes, mencionándoles cuáles son éstos.1 Además, es recomendable entregarles un informe de los hallazgos del estudio genético que debieran presentar cuando consulten un médico, para que éste lo considere al indicar un medicamento. Lo anterior debe completarse con la entrega de una lista de medicamentos seguros y no seguros de administrar a personas con porfirias de tipo agudo. Información adicional detallada sobre las porfirias agudas y remedios seguros y no seguros en pacientes con porfirias agudas, se encuentra, por ejemplo, en la página Web de Iniciativa Europea de Porfirias (European Porphyria Initiative, EPI) en http://www.porphyria-europe.com/.
AGRADECIMIENTOS
Este trabajo fue financiado parcialmente por proyecto No. 503 de la Fundación para Estudios Biomédicos Avanzados (FEBA), Facultad de Medicina, Universidad de Chile y parcialmente por proyecto No. A04155HS, GIS–Institut des Maladies rares: Network on rare diseases de la European Porphyria Initiative (EPI).
REFERENCIAS
1. Poblete–Gutiérrez P, Méndez M, Wiederholt T. et al. Diagnosis and treatment of the acute porphyrias: an interdisciplinary challenge. Skin Pharmacol Appl Skin Physiol 2001; 14: 393–400. [ Links ]
2. Badminton MN, Elder GH. Molecular mechanisms of dominant expression in porphyria. J Inherit Metab Dis 2005; 28: 277–86. [ Links ]
3. Armas R, Wolff C, Krause P, et al. The hepatic porphyrias: experience with 105 cases. Rev Med Chil 1992; 120: 259–66. [ Links ]
4. Kauppinen R. Porphyrias. Lancet 2005; 365: 241–52. [ Links ]
5. Sandberg S, Brun A, Skadberg O, Iversen IR, Benestad Y, Danielsen OK. Acute intermittent porphyria is a difficult diagnosis especially in children. Tidsskr Nor Laegeforen 2001; 121: 2822–5. [ Links ]
6. Norris PG, Elder GH, Hawk JL. Homozygous variegate porphyria: a case report. Br J Dermatol 1990; 122: 253–7. [ Links ]
7. Hift RJ, Meissner PN, Todd G, et al. Homozygous variegate porphyria: an evolving clinical syndrome. Postgrad Med J 1993; 69: 781–6. [ Links ]
8. Frank J, McGrath J, Lam H, et al. Homozygous variegate porphyria: identification of mutations on both alíeles of the protoporphyrinogen oxidase gene in a severely affected proband. J Invest Dermatol 1998; 110:452–5. [ Links ]
9. De Villiers PJN, Kotze MJ, Van Hereden CJ, et al. Overrepresentation of the founder PPOX gene mutation R59W in a South African patient with severe clinical manifestation of porphyria. Ex Dermatol 2005; 14: 50–5. [ Links ]
10. Frank J, Aita VM, Ahamad W, Lam H, Wolff C, Christiano AM. Identification of a founder mutation in the protoporphyrinogen oxidase gene in variegate porphyria patients from Chile. Hum Hered 2001; 51: 160–8. [ Links ]
11. da Silva V, Simonin D, Deybach JC, Puy H, Nordmann Y. Variegate porphyria: diagnostic value of fluorometric scanning of plasma porphyrins. Clin Chim Acta 1995; 238: 163–8. [ Links ]
12. Grandchamp B, Puy H, Lamoril J, Deybach JC, Nordmann Y. Review: molecular pathogenesis of hepatic acute porphyrias. Gastroenterol Hepatol 1996; 11: 1046–52. [ Links ]
13. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215. [ Links ]
14. Ganguly A, Rock MJ, Prockop DJ. Conformation–sensitive gel electrophoresis for rapid detection of single–base differences in double–stranded PCR products and DNA fragments: Evidence for solvent–induced bends in DNA heteroduplexes. Proc Nati Acad Sci USA 1993; 90: 10325–9. [ Links ]
15. Poblete–Gutiérrez P, Wolff C, Farias R, Frank J. A Chilean boy with severe photosensitivity and finger shortening: The first case of homozygous variegate porphyria in South America. Brit J Dermatol 2006; 154: 368–71. [ Links ]
16. Frank J, Jugert FK, Breitkopf C, Goerz G, Merk HF, Christiano AM. Recurrent missense mutation in the protoporphyrinogen oxidase gene underlies variegate porphyria. Am J Med Genet 1998; 79: 22–6. [ Links ]
17. Whatley SD, Puy H, Morgan RR. Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet 1999; 65: 984–94. [ Links ]
18. Frank J, McGrath JA, Poh–Fitzpatrick MB, Hawk JL, Christiano AM. Mutations in the translation initiation codon of the protoporphyrinogen oxidase gene underlie variegate porphyria. Clin Exp Dermatol 1999; 24: 296–301. [ Links ]
19. Deybach JC, Puy H, Robréau AM, et al. Mutations in the protoporphyrinogen oxidase gene in patients with variegate porphyria. Hum Mol Genet 1996; 5: 407–10. [ Links ]
20. Wiman A, Harper P, Floderus Y. Nine novel mutations in the protoporphyrinogen oxidase gene in Swedish families with variegate porphyria. Clin Genet 2003; 64: 122–30. [ Links ]
21. Bickers D, Frank J. The Porphyrias. In: Dermatology in general medicine. Fitzpatrick TB, Freedkey IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz ST (Eds.). 6a. Ed. New York: MacGraw Hill; 2003, p. 1435–66. [ Links ]
22. Hift RJ, Meissner PM. An analysis of 112 acute porphyric attacks in Cape Town, South Africa: evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore) 2005; 84: 48 – 60. [ Links ]
23. Moore MR, Hift RJ. Drugs in the acute porphyrias—toxicogenetic diseases. Cell Mol Biol (Noisy–le–grand) 1997; 43: 89–94. [ Links ] ]]>