Miofibromatosis infantil: reporte de un caso y revisión de la literatura
Infantile myofibromatosis: case report and literature review
María Letizia Fasola Maccari,1 Lisandro Manfrin,1 María Laura Galluzo,2 Adriana Vilma Scrigni1
1 Servicio de Clínica Médica
]]> 2 Servicio de Anatomía Patológica, Hospital Nacional de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Autor de correspondencia:
Dr. Lisandro Manfrin
Correo electrónico: lisandromanfrin@yahoo.com
Fecha de recepción: 11-02-10
Fecha de aceptación: 26-09-10
Resumen
]]> Introducción. La miofibromatosis infantil constituye una enfermedad caracterizada por la presencia (única o multicéntrica) de una neoplasia de naturaleza benigna. La etiología es desconocida. Predomina en varones menores de dos años. Se presenta clínicamente ya sea como un tumor solitario, único, que afecta piel, hueso, músculo o tejido celular subcutáneo en cabeza, cuello o tronco, con buen pronóstico; o bien en forma multicéntrica, con o sin compromiso visceral en corazón, pulmón, aparato gastrointestinal o riñón (de peor pronóstico). La confirmación requiere de una biopsia. El tratamiento puede ser conservador (control evolutivo), quirúrgico (cuando se presenta en forma solitaria) o con quimioterapia (en la forma visceral).Caso clínico. Se presenta aun paciente de 16 meses de vida con un tumor cervical en quien se confirmó el diagnóstico de miofibromatosis infantil; presenta una tumoración lateral izquierda de cuello. Se interna por dificultad respiratoria alta con estridor que requiere asistencia respiratoria mecánica. Es intervenido quirúrgicamente con resección completa de la tumoración y buena evolución posoperatoria.
Conclusiones. La miofibromatosis infantil debe considerarse en el diagnóstico diferencial de las neoplasias en la infancia temprana a pesar de su baja frecuencia. El tratamiento y el pronóstico dependerán de la localización y de la forma de presentación (solitaria o multicéntrica, con o sin compromiso visceral).
Palabras clave: miofibromatosis infantil, tumor cervical.
Abstract
Background. Infantile myofibromatosis (IM) is a disease characterized by solitary or multiple benign tumors. The etiology is unknown. IM isa benign mesenchymal disorder of early infancy and is more frequent in males. IM may present in two manners: asa solitary lesion most commonly in skin, bone, muscle, subcutaneous tissue, in head, neck and trunk, with good prognosis, or a multicentric form of IM with or without visceral involvement (heart, lung, gastrointestinal tract, kidney) with a poor prognosis. The definitive diagnosis of IM is confirmed by pathology. Treatment may be conservative (observation with close follow-up) or surgery (solitary form) or chemotherapy (visceral form).
Case report. We reporta case of a 16-months-old male patient with left neck tumor diagnosed as IM. He was admitted into intensive care unit because of respiratory distress with stridor caused by tumor compression. Mechanical ventilation was required by the patient who underwent surgery to resect the tumor. The patient had a favorable postoperative evolution.
Conclusions. IM must be considered in the differential diagnosis of tumors in early infancy, despite its low frequency. Treatment and prognosis depend on location, clinical form (solitary or multicentric), with or without visceral involvement.
Key words: infantile myofibromatosis, neck tumors.
]]>INTRODUCCIÓN
La miofibromatosis infantil (MI) constituye una enfermedad caracterizada por la presencia única o multicéntrica de una neoplasia de naturaleza benigna.1 Es una enfermedad de baja incidencia, aunque se considera como el tumor de tipo mesenquimático más frecuente en la etapa neonatal y en la primera infancia.2 La etiología es desconocida. Estos tumores son más frecuentes en niños menores de dos años. La presentación es esporádica o por herencia autosómica (recesiva, dominante o poligénica) de diversa expresión. Se trata de una entidad descrita por primera vez en 1954 por Stout3 e inicialmente conocida como "fibromatosis congénita generalizada". Desde la publicación de dicho trabajo varios casos fueron reportados en la literatura bajo diferentes nombres, incluyendo: fibromatosis congénita múltiple, hamartomas múltiples, leiomiomatosis múltiple vascular del recién nacido y fibromatosis múltiple congénita. En 1981 Chung y Enzinger realizaron una revisión de lo reportado hasta ese momento y delimitaron ciertas características que permanecen vigentes: introdujeron el nombre "miofibromatosis infantil" en lugar de fibromatosis congénita generalizada, establecieron la estirpe celular de la que proviene laneoplasiay señalaron el pronóstico en función de la localización y afectación: solitaria o multicéntrica, con o sin compromiso visceral.4 Finalmente, en 1989, Smith y sus colaboradores,5 así como Daimaru y sus colaboradores,6 acuñaron los términos "miofibroma" y "miofibromatosis". Esta denominación fue adoptada por la Organización Mundial de la Salud para describir a la forma solitaria (miofibroma) o multicéntrica (miofibromatosis).7
En el presente artículo se describe el caso de un paciente de 16 meses de edad que presentó un tumor cervical en el que se confirmó el diagnóstico de miofibroma infantil (MI). El objetivo es describir el caso clínico, su evolución y su tratamiento, así como revisar la literatura existente sobre esta entidad nosológica.
CASO CLÍNICO
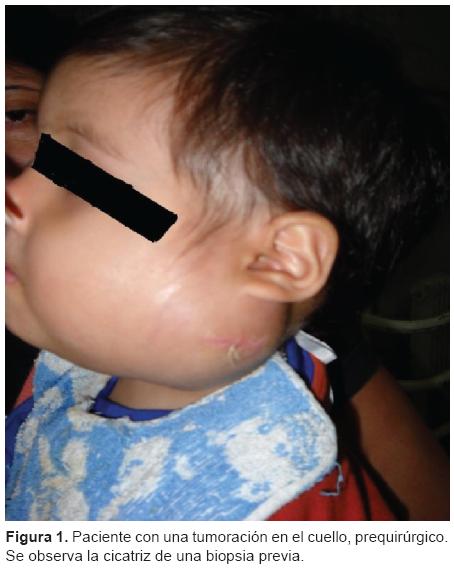
Niño de 16 meses de edad, sin antecedentes patológicos, que fue llevado a consulta a los 8 meses de edad en su ciudad de origen por presentar una tumoración lateral de cuello. En esa oportunidad se le diagnosticó proceso fibro-matoso por medio de una biopsia. Se internó de nuevo a los 16 meses por el aumento progresivo de la tumoración del cuello. En dicha ocasión presentó dificultad respiratoria con estridor inspiratorio y tiraje generalizado, por lo que requirió asistencia respiratoria mecánica (ARM) y fue derivado al Hospital Nacional de Pediatría Prof. Dr. Juan P. Garrahan. Ingresó a la Unidad de Cuidados Intensivos (UCI) con un cuadro clínico de insuficiencia respiratoria con compromiso de la vía aérea superior; se observó una tumoración laterocervical izquierda de aproximadamente 8 × 5 cm, renitente, adherida al plano subyacente, no dolorosa, sin signos locales de flogosis, que desplazaba el pabellón auricular hacia arriba, se extendía hasta el ángulo interno del ojo y por debajo del maxilar inferior sin atravesar la línea media, limitando la apertura bucal hasta 2.7 cm. No presentaba adenopatías ni visceromegalias (Figura 1). El paciente permaneció con ARM durante tres días y fue tratado con dexametasona a 0.6 mg/kg/día por el aumento del tamaño de la lesión y por los síntomas de la obstrucción de la vía aérea.
Se le realizó una tomografía axial computarizada (TAC) de cuello que mostró un tumor laterocervical izquierdo de aspecto lobulado que reforzaba de manera heterogénea con el contraste, con áreas hipodensas que podrían corresponder a zonas de necrosis. Se extendía desde la fosa pterigomaxilar hasta el plano de C5 y C6 y medía aproximadamente 5.8×7.3×4.2 cm. Dicha masa comprometía las regiones laterofaríngea izquierda, la retrofaríngea, la submaxilar y la parotidea. Se observaba el desplazamiento de la vía aérea y de las glándulas submaxilar y parótida homolateral con rarefacción de la interfase grasa y compresión de la vena yugular interna reducida a un trayecto filiforme. Se observaba la alteración de la cortical de la apófisis pterigoides lateral izquierda y la ausencia de extensión intracraneal. (Figuras 2 y 3).
]]>

En la TC de tórax se evidenció atelectasia del segmento apical del lóbulo superior derecho y de las subsegmentarias del segmento posterior del lóbulo superior derecho. Se realizó una ecografía abdominal que fue interpretada como normal. Luego de esta evaluación clínica y radiológica del paciente se programó una cirugía electiva que se realizó con la resección completa del tumor. El paciente presentó buena evolución posquirúrgica (Figura 4).

La pieza quirúrgica consistió en una formación nodular ovalada de bordes irregulares que midió 8.5 × 5 × 3 cm y pesó 100 g. La superficie externa era de coloración amarillenta-rojizay la superficie de corte era blanquecina uniforme y de aspecto arremolinado.
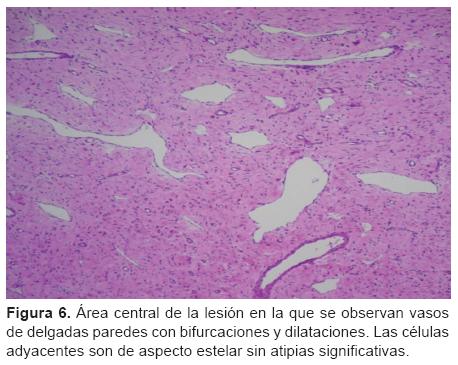
Histológicamente se evidenció una formación tumoral constituida por la proliferación de células ahusadas dispuestas en un patrón espiral irregular y surcada por vasos de paredes delgadas. En la región central, estos vasos dilatados y de fina pared presentaban frecuentes bifurcaciones y formas caprichosas, constituyendo un patrón hemangiopericitoide. Las células poseían citoplasmas eosinofilos y núcleos redondeados u ovoides con cromatina fina y de distribución uniforme, con ocasionales micro nucléolos evidentes. El tumor se encontraba revestido por una delgada capa de tejido conectivo, en sectores < 1 mm, constituyendo una pseudocápsula.
]]> En el centro de la lesión la proliferación neoplásica englobaba algunas fibras musculares esqueléticas residuales; a nivel periférico y extracapsular se evidenciaba tejido adiposo escaso maduro con vasos sanguíneos aislados de gran calibre. La inmunomarcación arrojó los siguientes resultados: vimentina, positivo; actina músculo liso, positivo; desmina, positivo focal; Ki67, bajo (menor a 5, lo que sugiere una cinética celular baja) (Figuras 5 y 6). Se llegó al diagnóstico de miofibroma/miofibromatosis infantil.
DISCUSIÓN
La MI es una proliferación fibroblástica a partir de células originadas en el tejido músculo-aponeurótico. Aunque su incidencia es baja es el tumor fibroso más frecuente en la infancia. Es una enfermedad pediátrica por excelencia dado que la mitad de los casos se presentan al nacimiento y, de estos, 90% se presenta antes de los 2 años de vida. Rara vez ocurre en niños mayores o en adultos.8
Si bien el origen es desconocido algunos autores consideran que estas lesiones presentan un patrón de herencia variable; otros consideran que pueden ser hamartomas y otros más asumen que son el resultado de una estimulación estrogénica intrauterina.9
]]> Existen tres formas de presentación clínica: la lesión única solitaria, las lesiones multicéntricas sin compromiso visceral y las lesiones multicéntricas con compromiso visceral.10La forma solitaria es el modo más común de presentación (50 a 75% de los casos), como lo señalan Chung y Enzinger4 y Muraoka y sus colaboradores,11 y esa fue la forma de presentación en nuestro paciente. Afecta especialmente la piel, el músculo, el hueso y el tejido celular subcutáneo en la cabeza, el cuello y el tronco. Esta variedad solitaria es más frecuente en varones (relación 2.4:1).12 Los nodulos únicos de miofibromas suelen ser firmes, bien circunscritos, no dolorosos y con una fase inicial de crecimiento rápido.13
La forma multicéntrica es menos frecuente (25 a 50% de los casos) y se clasifica en dos tipos: múltiple sin compromiso visceral y múltiple con compromiso visceral; afecta el corazón, los pulmones, el aparato gastrointestinal (hígado y páncreas) y los riñones.14
Los estudios radiológicos no sonpatognomónicos pero sirven para evaluar la extensión de la enfermedad (sobre todo para definir las formas multifocales), la progresión de la misma y el diagnóstico de recurrencia. Por eso, es importante realizar radiografías de huesos largos, tomo-grafías del cráneo y del tórax, ecografías abdominal y pelviana y ecocardiograma en todos los casos.15
En la literatura existe información sobre pacientes con compromiso visceral grave: mediastino,16 abdomen con afectacióndebazo,11 píloro,14 meso del colon transverso17 y dos casos con importante crecimiento tumoral intrauterino: un feto de 34 semanas y un neonato que muere al cuarto día de vida.2 El compromiso del sistema nervioso central es muy poco frecuente y se halla descrito en la bibliografía como la afectación exclusiva de la calota18,19 o bien como tumor de plexos coroideos,20 tumor de fosa posterior21 o también como compromiso de la duramadre y secundaria afectación de la calota.22
En todos los pacientes se requiere la confirmación por biopsia dado que, frente a un paciente como el que presentamos en este trabajo, se plantean diversos diagnósticos diferenciales: leiomioma, neurofibroma, sarcoma de partes blandas, neuroblastoma metastásico u otras fibromatosis, entre ellas el fibro sarcoma congénito infantil, el tumor miofibroblástico inflamatorio, la fibromatosis colli, la fibromatosis hialina o el trauma del músculo esternocleidomastoideo posparto.8
Histológicamente se trata de una proliferación de células formadoras de colágeno, ovoides a ahusadas, que muestran características imunohistoquímicas y electrónicas intermedias entre fibroblastos y células de músculo liso. Las células se agrupan en forma de nódulos, alternando áreas hipercelulares con otras hipocelulares que pueden tener necrosis central con o sin calcificación y generalmente con un patrón muy característico similar al hemangiopericitoma. En el pasado las lesiones con este patrón se clasificaban como hemangiopericitoma infantil; actualmente son reconocidas como parte del espectro de la MI y representan diferentes estadios de maduración de la misma entidad. Pueden estar bien circunscritas o infiltrar tejidos vecinos en forma digitiforme. Las determinaciones inmunohistoquímicas sugieren el fenotipo miofibroblástico, ya que son reactivas para marcadores como vimentina y actina músculo liso, mientras que la reactividad para la desmina es variable.
El tratamiento es expectante para la forma solitaria, con seguimiento clínico y por imágenes, dada la posibilidad de regresión espontánea. La forma multicéntrica requiere abordaje quirúrgico. También se considera oportuno el tratamiento quirúrgico cuando el tumor ocasiona compromiso clínico, como ocurrió con nuestro paciente que presentó insuficiencia respiratoria por el crecimiento del tumor en la vía aérea superior. La variante generalizada de MI es de mal pronóstico y se puede considerar el uso de quimioterapia.23,24
Después del tratamiento conservador (la evaluación periódica para determinar la regresión espontánea) o del quirúrgico se debe realizar el seguimiento de estos pacientes porque existe la posibilidad de recurrencia. Ésta se presenta cercana a 5% para la forma solitaria. También existe la posibilidad de recidiva en el caso de exéresis incompleta.
El pronóstico de la enfermedad depende del modo de presentación. Habitualmente es benigno, con regresión espontánea total en un periodo de 1 a 2 años para la variante solitaria e, incluso, para la variante multifocal sin compromiso visceral. La forma multicéntrica con compromiso gastrointestinal, cardiaco o pulmonar puede presentar una mortalidad de hasta 73%.14
]]> La MI debe considerarse para el diagnóstico diferencial de las neoplasias en la infancia temprana a pesar de su baja frecuencia. El tratamiento y el pronóstico dependerán de su localización y su forma de presentación: solitaria o multicéntrica, con o sin compromiso visceral.
REFERENCIAS
1. Scheper MA, DiFabio VE, Sauk JJ, Nikitakis NG. Myofibroma-tosis: a case report with a unique clinical presentation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2005;99:325-330. [ Links ]
2. Pelluard-Nehmé F, Coatleven F, Carles D, Alberti EM, Briex M, Dallay D. Multicentric infantile myofibromatosis: two perinatal cases. Eur J Pediatr 2007; 166:997-1001. [ Links ]
3. Stout AP. Juvenile fibromatoses. Cancer 1954;7:953-978. [ Links ]
4. Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer 1981;48:1807-1818. [ Links ]
5. Smith KJ, Skelton HG, Barrett TL, Lupton GP, Graham JH. Cutaneous myofibroma. Mod Pathol 1989;2:603-609. [ Links ]
6. Daimaru Y, Hashimoto H, Enjoji M. Myofibromatosis in adults (adult counterpart of infantile myofibromatosis). Am J Surg Pathol 1989;13:859-865. [ Links ]
7. Rubin BP, Bridge JA. Myofibroma/Myofibromatosis. En: Fletcher CDM, Unni KK, Mertens F, eds. World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon: IARC Press; 2002. pp. 59-61. Disponible en: http://www.iarc.fr/en/publications/pdfs-online/pat-gen/bb5/BB5.pdf [ Links ]
8. Franzese CB, Carrón J. Infantile myofibromatosis: unusual diagnosis in an older child. Int J Pediatr Otorhinolaryngol 2005;69:865-868. [ Links ]
9. Kaplan SS, Ojemann JG, Grange DK, Fuller C, Park TS. Intracranial infantile myofibromatosis with intraparenchymal involvement. Pediatr Neurosurg 2002;36;214-217. [ Links ]
]]>10. Wiswell TE, Davis J, Cunningham BE, Solenberger R, Thomas PJ. Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 1988;23:314-318. [ Links ]
11. Muraoka I, Ohno Y, Kamitamari A, Okada M, Moriuchi H, Kanematsu T. Congenital occurrence of solitary infantile myofibromatosis of the spleen. J Pediatr Surg 2008;43:227-230. [ Links ]
12. Stanford D, Rogers M. Dermatological presentations of infantile myofibromatosis: a review of 27 cases. Australas J Derm 2000;41:156-161. [ Links ]
13. Prado Calleros HM, Castillo Ventura BB, Arrieta Gómez JR, García García MA, Sánchez Marín LA, et al. Tumores fibrosos/ miofibrosos en cabeza y cuello. Rev Hosp Gral Dr. M Gea González 2006;7:76-81. [ Links ]
14. Rohrer K, Murphy R, Thresher C, Jacir N, Bergman K. Infantile myofibromatosis: a most unusual cause of gastric outlet obstruction. Pediatr Radiol 2005;35:808-811. [ Links ]
]]>15. Spadola L, Anooshiravani M, Sayegh Y, Jéquier S, Hanquinet S. Generalised infantile myofibromatosis with intracranial involvement: imaging findings in a newborn. Pediatr Radiol 2002;32:872-874. [ Links ]
16. Short M, Dramis A, Ramani P, Parikh DH. Mediastinal and pulmonary infantile myofibromatosis: an unusual surgical presentation. J Pediatr Surg 2008;43:e29-e31. [ Links ]
17. Hermida-Pérez JA, Ochoa-Montes de Oca J, Ochoa-Urdanga-rarain O, Bastian-Manso L, Morell-Molina E. Miofibromatosis infantil. Su relación con el aparato genitourinario. Presentación de un caso y revisión de la literatura. Arch Esp Urol 2007;60:571-576. [ Links ]
18. Okay O, Kulacoglu S, Ergungor MF, Secer M, Han O, Dalgic A. An unusual mesenchymal tumor of the skull: infantile myofibromatosis. Pediatr Neurosurg 2008;44:178-180. [ Links ]
19. Rossbach C, Tannapfel A, Troebs RB, Hirsh W, Koerholz D. Successful treatment of relapsed multifocal nonvisceral infantile myofibromatosis. Pediatr Hematol Oncol 2005;22:695-698. [ Links ]
]]>20. Hyun Yoo J, Naeini RM, Hunter JV. Infantile myofibromatosis involving the choroid plexus. Pediatr Radiol 2008;38:107-110. [ Links ]
21. Chapman P, Judd CD, Felgenhauer JL, Gruber DP, Mornin D. Infantile myofibromatosis of the posterior fossa. Am J Roent-genol 2005;184:1310-1312. [ Links ]
22. Tamburrini G, Gessi M, Colosimo C Jr, Lauriola L, Giangaspero F, Di Rocco C. Infantile myofibromatosis of the central nervous system. Childs Nerv Syst 2003; 19:650-654. [ Links ]
23. Day M, Edwards AO, Weinberg A, Leavey PJ. Brief report: successful therapy of a patient with infantile generalized myofibromatosis. Med Pediatr Oncol 2002;38:371-373. [ Links ]
24. Williams W, Craver RD, Correa H, Velez M, Gardner RV. Use of 2-chlorodeoxyadenosine to treat infantile myofibromatosis. J Pediatr Hematol Oncol 2002;24:59-63. [ Links ]
]]>