nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of conditions that usually start as a reversibly benign simple hepatic steatosis.
However, it can progress to non-alcoholic steatohepatitis (NASH), cirrhosis, and finally hepatocellular carcinoma.1,2
NAFLD development is associated with risk factors such as diet (high in saturated fat and processed meat), altered metabolism, metabolic liver disease, and metabolic syndrome.3-5
It is clear that with population aging and the current metabolic syndrome epidemy NAFLD’s clinical and economic burden will undoubtedly be overwhelming in the coming decades worldwide.6
This article will address the association between NAFLD, metabolic syndrome, and cardiovascular disease.
NAFLD, a silent pandemic. The global prevalence of NAFLD is 25.4%, being higher in South America (30.45%) and the Middle East (31.79%) and lower in Africa (13.48%). In Asia and the Pacific, NAFLD numbers are also increasing, mainly due to changes in diet and urbanization, and the adoption of the Western lifestyle.1
The growth of metabolic diseases such as obesity and type 2 diabetes mellitus (DM2) parallels the increase in NAFLD. The prevalence of NAFLD is between 50% and 75% in people with DM2 and between 80% and 90% in obese subjects; while in patients who have more than one risk factor, the incidence is almost total (80-100%); Similar Figures have been observed in the population waiting for a bariatric surgery procedure.4,7
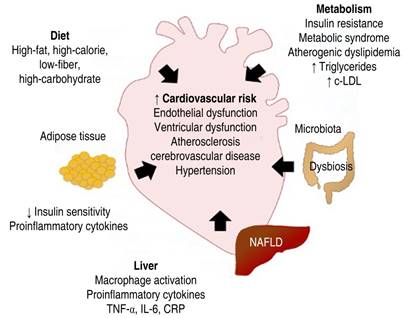
Pathogenesis of NAFLD. The exact mechanism(s) establishing fat accumulation in the hepatocyte, especially those that set the development of inflammation, fibrosis, and the evolution of the disease, are not precisely known. The pathology is complex and multifactorial since it involves multiple genetic, metabolic, environmental, and nutritional factors.7 Various theories have emerged to describe the pathophysiology of NAFLD. The first was the two-hit hypothesis. The first hit is caused by an excessive accumulation of lipids in the liver, produced by multiple factors, such as hypercaloric diets, sedentary lifestyle, obesity, and insulin resistance (IR). This first blow sensitizes the liver. The second hit is secondary to various metabolic insults that lead to inflammation and fibrogenesis. This theory was considered too simple to explain the pathogenesis of NAFLD and, as a consequence of the multiple hit impact hypothesis was developed; this hypothesis states that environmental factors such as excessive high-calorie diets and low physical activity, and genetic factors can favor the development of insulin resistance, obesity, adipose tissue dysfunction, and alterations in the intestinal microbiota. Intercellular crosstalk between hepatocytes, Kupffer cells, and hepatic stellate cells is also involved in the pathogenesis of NAFLD.8 All together are involved in the development and progression of the disease.
Interrelation of NAFLD and metabolic syndrome. Insulin resistance and NAFLD. Insulin resistance (IR) is a weak or altered biological response to insulin, decreasing insulin-mediated glucose uptake, despite normal or elevated insulin concentrations. In states of IR, the pancreas releases more insulin to solve the defects in peripheral glucose uptake and eliminate hepatic glucose production. In hepatocytes, insulin promotes glycogenesis, inhibits gluconeogenesis, and activates de novo lipogenesis (DNL), that is, the formation of triglycerides (TG) from glycerol and three molecules of free fatty acids (FFA). Contrariwise, IR increases lipolysis (the catabolic release of FFA from TG), raising their blood concentration. The accumulation of FFA and lipid metabolites in hepatocytes activates kinases, which phosphorylate serine or threonine residues of the receptor, altering the normal phosphorylation of tyrosine residues and leading to deficient insulin signaling pathway and causing the phenomenon known as IR. The altered signaling cascade interferes with the translocation of the GLUT-4 glucose transporter to the cell membrane, impeding the glucose inflow from the blood and increasing the level of glycemia. Furthermore, IR causes a decrease in glycogenesis and stimulates gluconeogenesis. Probably, the hyperinsulinemia that accompanies IR, while the pancreas can secrete insulin, is a primary defect more than a compensation mechanism of IR.9 Although insulin promotes DNL in hepatocytes, hepatic IR does not lead to suppression of DNL but to an increase through mechanisms that are not completely clear.10-13
Association between NAFLD, obesity, dyslipidemias, and adipose tissue dysfunction. In obesity, the excessive accumulation of lipids in adipocytes increases the size of fat cells and the entire mass of adipose tissue (AT). Not in all, but in most obesity cases, adipocyte function is impaired. The activation of c-JNK and lkKbeta-dependent inflammation pathways causes reduced insulin sensitivity. Obesity is a risk factor for IR and NAFLD. IR in adipose tissue alters the antilipolytic effect of insulin, increasing lipolysis. The hyperinsulinemia and hepatic IR induced increases in hepatic DNL and a more significant release of TG from the liver, which translates into a high load of circulating lipids delivered to the AT, aggravating the adipocyte’s functional deficiency. The AT plays an essential endocrine role in the body. It secretes hormones and adipokines such as adiponectin and leptin, critical metabolic regulators. AT functional alterations are related to the development and progression of NAFLD and NASH. Adiponectin improves hepatic IR since it suppresses glycogenolysis and lipogenesis, improving glucose utilization. Its deficiency could intervene in mitochondrial dysfunction, IR, and obesity. Adiponectin levels decrease as adipocyte size and IR increase and relate to the low plasma concentrations found in subjects with NAFLD, which predicts the risk of NASH. On the other hand, leptin acts centrally, reducing food intake and increasing energy use, preventing the accumulation of lipids in organs other than the AT, such as the liver. Obese subjects and those with NASH develop leptin resistance, increasing plasma concentrations. The increase in leptin relates to decreased glucose uptake and increased gluconeogenesis, generating hyperglycemia, and therefore participating in the development of IR.14-16 Finally, increased lipogenesis, resulting from an increased fat mass that is responsible for the high levels of circulating FFA, overproduction of VLDL, and other abnormalities in lipid metabolism, can lead to significant lipid anomaly characterized by an increase in the population of small and dense LDL, the concentration of TG and a decrease in HDL (hypoalphalipoproteinemia).8 This atherogenic dyslipidemia or lipid triad is seen mainly in dysmetabolic, diabetic or not, obese or overweighed patients and seems to be the primary lipid abnormality causing myocardial infarction in Mexicans.17
Relationship between NAFLD, cardiovascular diseases, and hypertension. There is diverse evidence suggesting that patients with NAFLD have a high cardiovascular risk, representing the leading cause of death in these patients.18,19 Several epidemiological and clinical studies have suggested a role for NAFLD in the progression of different cardiovascular manifestations, such as endothelial dysfunction, left ventricular dysfunction, atherosclerotic cerebrovascular disease (CVD), abnormalities of the cardiac conduction system, and ischemic stroke.20,21 There are multiple pieces of evidence that NAFLD is a promoter risk factor for the occurrence of high blood pressure (HBP), and at the same time, HBP can aggravate the fibrotic reaction in fatty livers. The mechanisms of this back-and-forth relationship are not entirely elucidated. Still, everything indicates that inflammation, endothelial dysfunction, increased oxidative stress, gut microbiota abnormalities, and overexpression of the renin-angiotensin-aldosterone system, among many other phenomena, can intertwine both clinical conditions.22,23 The mechanisms by which NAFLD increases CVD risk are complex and involve various pathways at different functional and structural levels, such as metabolism, the cardiovascular system, and liver function.24-26 The liver has the most significant number of resident macrophages and an increase in pro-inflammatory cytokines (TNF-α, IL-6, CRP) that can be chronically released into the circulation and promote chronic inflammation and thrombotic susceptibility.27,28 Therefore, it is considered that liver damage and the release of proinflammatory cytokines result in systemic microvascular damage, abnormalities of the coagulation system, altered endothelial dysfunction, and the generation of oxidative stress. It is also suggested that NAFLD and CVD share a common hereditary predisposition. The endothelial damage induces an increase in arterial stiffness, which promotes the development of hypertension and CVD.25,26 High serum concentrations of vascular endothelial growth factor (VEGF) and high levels of prothrombotic factors (factors VIII, IX, XI, and XII) have been reported in patients with NAFLD, and they correlate with hepatic fat content. Therefore, they may be related to an increased cardiovascular risk. Recently, it has been reported that there is a systemic role for liver tissue-specific molecules (hepatokines), which appear to affect multiple metabolic pathways. These molecules play a relevant role in developing cardiovascular complications in patients with NAFLD. For example, the fibroblast growth factor 21 (FGF-21), a peptide secreted by the liver, negatively affects the cardiovascular system. Increased serum concentrations of FGF-21 are associated with thickening of the carotid intima-media and coronary atherosclerosis.29-32 Therefore, NAFLD stimulates pro-inflammatory and prothrombotic factors, contributing to several chronic diseases, including ischemic heart disease, cardiomyopathy, cardiac arrhythmias, and chronic kidney disease.2
Metabolic syndrome and NAFLD. Metabolic syndrome (MS) or dysmetabolic obesity is a set of metabolic disorders consisting of centrally distributed obesity, decreased concentrations of cholesterol bound to high-density lipoproteins (HDL), elevated concentrations of TG, high blood pressure arterial hypertension, and hyperglycemia. All these alterations are present in patients with NAFLD. The prevalence of MS in obese patients with NAFLD is 67%. Furthermore, the presence of MS was associated with an increased risk of NASH and severe fibrosis. However, not all patients with NAFLD exhibit the typical features of MS. Approximately 30% of NAFLD patients suffer from metabolic abnormalities.33,34 In many patients, both clinical conditions, NAFLD and MS, are early, almost simultaneous manifestations of IR. The current concept signals that NAFLD is the hepatic expression of MS, but frequently, hepatic abnormalities can worsen IR. On the contrary, the lipid abnormalities were seen in MS; for example, the so-called lipid triad can aggravate the lipid liver storage.35
Microbiota and NAFLD. Alterations in the microbiota (dysbiosis) have recently been associated as part of the origin of the pathogenesis of NAFLD; for example, a greater abundance of Escherichia coli and Bacteroides vulgatus has been found in the fecal microbiota of patients with NAFLD. An increase in the intestinal barrier permeability has also been found with hypercaloric diets high in saturated fat, low in fiber, rich in refined carbohydrates, and high in fructose in patients with NAFLD. Dysfunction of the intestinal barrier allows the activation of receptors that trigger the production of proinflammatory cytokines, such as TNF-α. Also, it will enable the passage of lipopolysaccharides (LPS) into the circulation, activating the immune system, including Kupffer cells (hepatic macrophages), triggering more inflammatory processes, oxidative stress, and liver damage.36
Conclusion
NAFLD is a growing problem for public health in most countries worldwide. Uncertain diagnosis without specific treatment is one of the significant challenges of basic science and clinical science today. Considering the risk factors for the development of NAFLD (Figure 1), we can justify the need for comprehensive management of the patient from the early stages, from identifying and controlling overweight or obesity to preventing dyslipidemia, metabolic syndrome, and other complications. The central pillar in public health continues to be promoting a healthier lifestyle, including a diet with a low content of simple carbohydrates and saturated fat, rich in polyunsaturated omega-3 fat acids, fiber, prebiotics, probiotics, and nutraceuticals such as epicatechin from cocoa. In addition, an increase in physical activity reduces the impact of Western life on the liver and cardiovascular health.