











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El feocromocitoma es un tumor que se origina a partir de las células cromafines de la médula suprarrenal, que produce, almacena y secreta catecolaminas; clínicamente los pacientes se presentan con síntomas por el exceso de catecolaminas (hipertensión arterial persistente o paroxística), en ocasiones con consecuencias graves. Este tumor puede asociarse a otros síndromes clínicos hereditarios y es potencialmente curable con el tratamiento quirúrgico1,2.
El término feocromocitoma proviene del griego: phios significa oscuro, chromo significa color y cytoma, tumor. Se refiere al color que adquieren las células cuando se tiñen con las sales de cromo1,2.
Los antecedentes históricos de esta entidad datan de 1522, cuando Eustaquio descubre las glándulas suprarrenales. Informa su hallazgo en 1563. Frankel, en 1886, informa el primer caso de un feocromocitoma bilateral. Neusser, por primera vez, asocia el feocromocitoma con la hipertensión arterial, Labbé y otros, en 1892, lo relacionan con la hipertensión arterial paroxística. Suzuki, en 1910, reconoce por primera vez la asociación del feocromocitoma y la enfermedad de von Recklinghausen. Roux (Suiza) en 1926 y Mayo (EE.UU.), también en 1926, e independientemente, reportan la primera exéresis de un feocromocitoma con éxito. Ninguno de los dos hizo el diagnóstico preoperatorio. Kvale, et al., en 1956, establecen el tratamiento preoperatorio para controlar la presión arterial y poder operar a estos pacientes1,3,4.
Los feocromocitomas son tumores encapsulados total o parcialmente, muy vascularizados; generalmente tienen un diámetro mayor de 3 cm, con una media entre 5-6 cm en el momento del diagnóstico; pesan entre 50 y 200 g, aunque existen más grandes. En el corte de sección la superficie tiene color marrón. Los casos hereditarios, por lo general, son bilaterales y los esporádicos unilaterales3,5,6.
Se ha descrito para este tumor la regla de los 10, que hace referencia a: el 10% son extraadrenales, el 10% se presentan en niños, el 10% son múltiples o bilaterales, el 10% recidiva luego de la cirugía, el 10% son malignos y el 10% son familiares4-6.
En este artículo se presenta un paciente atendido en el Servicio de Oncología Clínica del Hospital Clínico-Quirúrgico Hermanos Ameijeiras con diagnóstico de feocromocitoma maligno extraadrenal.
Caso clínico
Paciente de sexo masculino de 40 años, raza blanca, con antecedentes patológicos personales de un feocromocitoma a los 11 años de edad constatado en estudios imagenológicos al paciente, por presentar cifras elevadas de tensión arterial y cefaleas recurrentes. Se le realiza una nefrectomía derecha, con conservación de la glándula suprarrenal correspondiente, y se diagnóstica un feocromocitoma de aproximadamente 6 cm en su eje mayor. El paciente se mantiene en remisión de su enfermedad y asintomático con controles semestrales y posteriormente anuales. Sin otros antecedentes patológicos familiares ni alergias medicamentosas.
Refiere que, desde hace aproximadamente ocho meses antes de la consulta, comenzó con un dolor retroesternal, en ocasiones punzante, que se exacerbaba con los cambios de posición; posteriormente notó un aumento de volumen en la región anterior del tórax. Recibe tratamiento con antiinflamatorios no esteroideos y relajantes musculares con alivio parcial.
Por este motivo se le realizan estudios imagenológicos en otro centro de salud (radiografía de tórax, ecografía abdominal y tomografía computarizada [TC]), observándose imagen osteolítica que destruye dos tercios superiores del esternón. Se realiza curetaje y biopsia y se diagnóstica metástasis de un feocromocitoma extraadrenal y es remitido a nuestra institución.
Se recoge que consumía dos tazas de café diarias, no fuma ni ingiere bebidas alcohólicas.
Al examen físico se constata índice de masa corporal de 26.3 kg/m2. Se constata aumento de volumen de la pared anterior del tórax sin cambios de coloración de la piel comprometida, palpándose a la altura del manubrio esternal una tumoración de aproximadamente 4,5 cm de diámetro mayor, de consistencia dura y ligeramente dolorosa a la palpación, de varios meses de evolución. No existen otras lesiones cutáneas ni se palpan adenopatías en cadenas ganglionares accesibles. Además, se constataron cifras elevadas de tensión arterial (130/90 mmHg) y frecuencia cardiaca con tendencia a la taquicardia (98-110 latidos por minuto). El resto de la exploración física fue normal.
Al evaluar la Eastern Cooperative Oncology Group (ECOG), fue de 0.
Los exámenes de laboratorio (21/11/17) mostraron:
- Hematocrito: 0.35%.
- Hemoglobina: 12.3 g/l.
- Velocidad de sedimentación globular: 95 mm/h.
- Leucocitos: 11.1 x 109 x l.
- Glucemia: 5.1 mmol/l.
- Gamma glutamil transpeptidasa: 63 u/l.
- Fosfatasa alcalina: 345 u/l.
- Creatinina: 108 mmol.
- Acido úrico: 440 mmol/l.
- Colesterol: 6.1 mmol/l.
- Triglicéridos 1.8 mmol/l.
- Aspartato aminotransferasa: 90 u/l.
- Alanina aminotransferasa: 59 u/l.
- Antígeno carcinoembrionario: 0.2 ng/ml.
- Enolasa neuronal específica: 13.40 pg/ml.
Exámenes imagenológicos:
- Ecografía abdominal: hígado de tamaño y ecogenicidad normal sin demostrar lesión nodular, no se encuentran otras alteraciones del hemiabdomen superior, se observa el riñón izquierdo único y compensador con pequeño quiste hacia el polo inferior.
- Radiografía de tórax: se aprecia área de osteólisis en proyección a la parte superior del esternón. Resto del informe sin otras alteraciones pleuropulmonares.
-
- TC (EV) de tórax y abdomen (18/10/17): una lesión expansiva y ovoide de bordes irregulares (Figs. 1 y 2), que provoca insuflación y rompe la cortical ósea de gran parte del esternón (manubrio esternal); tiene aspecto tumoral y mide 58 x 50 mm, densidad heterogénea (entre 20 y 40 UH), con amplia osteólisis del esternón que destruye los 2/3 superiores y ocupa el espacio o compartimento mediastinal anterior, abombando la pared anterior y englobando partes blandas adyacentes.
A nivel pulmonar se observa pequeña calcificación pleural anterior derecha sin derrame. Pequeña adenopatía pretraqueal derecha de 8 x 4 mm de aspecto inflamatoria. En abdomen no se observan alteraciones de sus estructuras y órganos, se constata el riñón izquierdo único y compensador, con pequeño quiste de 8 mm hacia el polo inferior. Ambas glándulas suprarrenales presentes y normales. Sin lesiones focales, algunas adenopatías menores de 1cm, no ascitis.
- Tomografía por emisión de positrones-tomografía computarizada (12/01/18): estudio compatible con tumoraciones malignas óseas a nivel del cráneo en su parte superior (ambos parietales a nivel de la sutura sagital), en la parte superior del esternón (manubrio), a nivel del cuerpo del 1.er hueso costal izquierdo y el cuerpo de la 2.ª vértebra cervical, con captación patológica del radioisótopo (Galio68) en los sitios mencionados (Figs. 3 y 4).
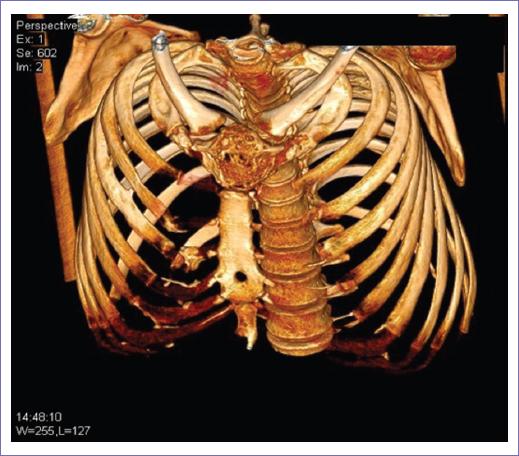
Figura 1 Se muestra la lesión focal osteoblástica metastásica de un feocromocitoma con insuflación y ruptura de la cortical ósea de gran parte del esternón (manubrio esternal) (vista frontal).

Figura 2 Corte axial que muestra la lesión metastásica de un feocromocitoma maligno extraadrenal en la parte superior del esternón.
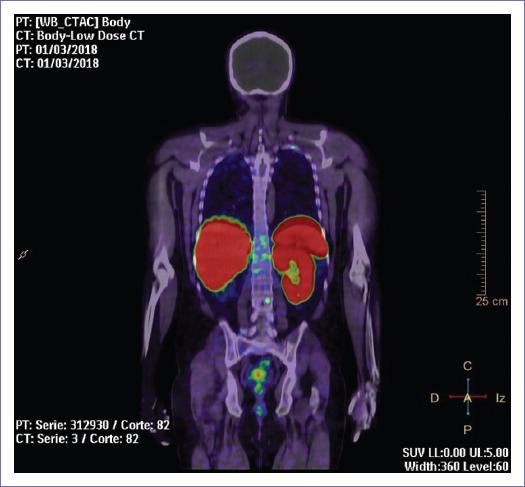
Figura 3 Corte coronal que muestra la captación patológica del radiofármaco en el estudio con tomografía por emisión de positrones-tomografía computarizada (Galio68) de una lesión metastásica por feocromocitoma maligno extraadrenal en la porción media del cuerpo del 1.er hueso costal izquierdo.

Figura 4 Corte sagital que muestra la captación patológica del radiofármaco en el estudio con tomografía por emisión de positrones-tomografía computarizada (Galio68) de una lesión metastásica por feocromocitoma maligno extraadrenal en la parte superior de los huesos parietales y parte superior del esternón.
Se realizó curetaje de la lesión del esternón y biopsia: infiltración ósea por feocromocitoma metastásico. El estudio inmunológico usando la técnica de inmunohistoquímica mostró: cromogranina (+) y Ki-67 < 10% de los núcleos tumorales.
Actualmente el paciente está clínicamente estable, en tratamiento con opiáceos débiles y citado en Oncología Médica, pendiente de tratamiento sistémico con metayodobencilguanidina.
Discusión
El feocromocitoma es un tumor neuroendocrino raro asociado a una morbilidad y mortalidad elevada. Aunque se trata de una enfermedad rara, con incidencia anual de 2 a 8 casos por 1,000,000 de habitantes y una prevalencia del 0.1 al 0.6% en pacientes con hipertensión el manejo temprano, representa una oportunidad de curación y evitación de consecuencias a largo plazo7.
Por lo general en estos tumores predomina la localización adrenal (90% de los casos), entre la 4.ª y 5.ª décadas de la vida, con mayor afectación del sexo femenino. Los casos extraadrenales (10-20%) se hallan con más frecuencia en pacientes jóvenes, entre la 2.ª y 3.ª décadas, sin predominio de sexo. De estos el 85% son infradiafragmáticos y el 10% torácicos2,8.
El paciente es atendido en el Servicio de Oncología Clínica del Hospital Clínico-Quirúrgico Hermanos Ameijeiras, con diagnóstico de feocromocitoma maligno extraadrenal pertenece a ese 10% que refiere la literatura científica, siendo operado de ese tumor en la infancia a la edad de los 11 años, lo que hace más raro el caso al revisar la bibliografía consultada, que expone mayor incidencia en la 2.ª y 3.ª décadas de la vida.
Tradicionalmente se ha establecido que un 10% de los feocromocitomas son extraadrenales. Al analizar los datos reportados en diferentes series, observamos que no se cumple exactamente esta proporción, oscilando entre el 10 y el 42%. También es conocido que un 10% de los tumores derivados del tejido cromafín son malignos y que estos se hallan en mayor proporción en los de localización extradrenal9.
Otros autores establecen que la angiogénesis tumoral puede ser útil para determinar las probabilidades de desarrollar enfermedad maligna. Para ORiordain, et al. los tumores extraadrenales funcionantes mayores de 5 cm de diámetro y la presencia de metástasis a distancia, descritas hasta 15 años después de la resección quirúrgica del tumor primario, tienen más incidencia de enfermedad maligna. En otra serie no se observa mayor supervivencia entre tumores funcionantes y no funcionantes9,10.
El diagnóstico de un feocromocitoma maligno se constató por tratarse de una localización extraadrenal, con un tamaño del tumor de 6 cm y la aparición de metástasis a distancia después de varios años de operado, aunque en el caso muestra metástasis después de 29 años de la resección quirúrgica del tumor primario, que no es lo más usual según los planteamientos anteriores.
En el caso del feocromocitoma, los determinantes principales en esta nueva clasificación son el tamaño del tumor, además de la presencia y localización de las metástasis. En términos generales, un tumor mayor de 5 cm tiene peor supervivencia y más riesgo de malignidad que un tumor pequeño. Los paragangliomas también tienen peor supervivencia y mayores tasas de malignidad. La localización del tumor predice mejor la agresividad que el tamaño. Mientras que solo un 3% de los feocromocitomas menores de 5 cm tienen metástasis, los paragangliomas menores de 5 cm tienen compromiso a distancia hasta en el 20% de los casos. Una debilidad de esta clasificación es que no tiene en cuenta los aspectos genéticos y es posible que las mutaciones en SDHB tengan un peor pronóstico, como se ha sugerido en algunos estudios. Sin embargo, pacientes con enfermedad aparentemente esporádica pueden tener un comportamiento clínico muy agresivo. Según se defina con claridad el pronóstico y se valide la clasificación, se tendrá en cuenta el genotipo del tumor. El tipo de compromiso metastásico también determina el pronóstico. Los pacientes con metástasis óseas solas tienen mejor pronóstico que aquellos que la poseen en hígado y pulmones2,11,12.
Contrario a lo que sucede en otros tipos de cáncer, no hay marcadores tumorales o histológicos que distingan con precisión los tumores malignos de los benignos. La definición aceptada de malignidad a la fecha, de acuerdo con la Organización Mundial de la Salud, es la presencia de metástasis, a pesar de que se han propuesto diferentes marcadores de agresividad, como la elevación de la proteína Ki-67, la presencia de necrosis, la invasión vascular, la ruptura de la cápsula, el alto número de mitosis y el tamaño tumoral. La tasa de malignidad clásicamente se ha considerado alrededor del 10%; sin embargo, la realidad actual sitúa la frecuencia del feocromocitoma maligno cercana al 26%, incluso, la experiencia del MD Anderson Cáncer Center en EE.UU. arroja que el 17% de estos tumores son malignos12,13.
Desde el punto de vista del seguimiento, un problema importante es que hasta el 50% de los feocromocitomas y paragangliomas malignos se clasifican inicialmente como benignos y no se realiza ningún tipo de seguimiento luego del manejo quirúrgico del tumor primario. Desafortunadamente, algunos de estos casos se presentan tardíamente con metástasis que carecen de opción quirúrgica y en los cuales la quimioterapia tiene una eficacia variable. Por tal razón, se recomienda que se realice genotipificación para el gen SDHB en todos los pacientes con feocromocitoma maligno, dado que se asocia frecuentemente con paragangliomas simpáticos malignos. El tamaño del tumor primario y su localización son predictores clínicos de metástasis y de supervivencia en los pacientes con feocromocitoma y paraganglioma. La localización del tumor primario es un predictor más fuerte que el tamaño del tumor. Actualmente, los únicos marcadores clínicos reconocidos de metástasis, supervivencia y pronóstico en pacientes con esta enfermedad son la presencia de mutaciones germinales del SHDB, un tumor primario mayor que 5 cm y la localización simpática extraadrenal (paraganglioma). En caso de presentarse tumores con estas características se puede necesitar una aproximación quirúrgica más agresiva y un seguimiento a largo plazo2,13.
La malignidad del tumor se establece únicamente ante la presencia de metástasis a distancia, en sitios donde normalmente no hay tejido de paraganglia (p. ej., nódulos linfáticos, hígado, pulmones y hueso)1,13,14.
Las vértebras constituyen una localización atípica de la diseminación a distancia de los feocromocitomas. Las metástasis deben ser resecadas si es posible. El dolor por las metástasis óseas puede ser tratado con radioterapia externa. La ablación por radiofrecuencia de las metástasis hepáticas y óseas puede ser definitiva en pacientes seleccionados. En tumores agresivos con afectación de la calidad de vida puede considerarse la quimioterapia5,14.
La extirpación de la metástasis vertebral, facilitada por la embolización previa por métodos de radiología intervencionista, que minimiza la morbilidad operatoria, puede suponer la curación de la enfermedad. No obstante, es preciso un seguimiento exhaustivo7,10,14.
Conclusiones
Este paciente presentó un feocromocitoma operado hace 29 años y en estudios de extensión para su evaluación se constataron metástasis óseas a distancias, lo que muestra la malignidad del tumor, reforzado este criterio por el tamaño de este. Las metástasis extraadrenales del caso descritas en diferentes sitios (a nivel del cuerpo del 1.er hueso costal izquierdo y el cuerpo de la 2.a vértebra cervical, huesos de la bóveda craneana [región superior de los parietales] y la porción superior del esternón) evidencian el comportamiento atípico y raro de la diseminación del feocromocitoma en este paciente. No se encontraron publicaciones de un caso similar al reportado en la literatura revisada sobre el tema.
El feocromocitoma es una enfermedad poco común que ha llamado la atención a investigadores y clínicos, que a lo largo de los años han colaborado para establecer métodos más efectivos de su detección, localización y manejo; no obstante, su tratamiento sigue siendo un auténtico reto para la medicina actual.

