











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Turner syndrome (TS) is defined as the combination of a series of characteristic physical findings and the total or partial absence of an X chromosome. It is one of the most frequent chromosomal abnormalities. It affects approximately 3% of all female fetuses and has an approximate incidence of 1 per 2500 newborn women. 50-60% of the cases demonstrate a monosomy or complete absence of X chromosome (45 X), and in most of the times, the lost X chromosome is of paternal origin. In 20% of cases, structural alterations are found in X chromosome (partial monosomy) as major deletions, microdeletions, isochromosomes, or ring chromosome. The remaining 20% are patients with the presence of two or more cell lines derived from the same zygote (mosaicism), and in at least one of them, there are numerical or structural alterations of an X chromosome.
The clinical spectrum of TS is very variable. Although no isolated finding is specific, they confer a very characteristic phenotype as a whole, among the characteristics are anomalies in the development of the bones of the face: retrognathia, micrognathia, and underdeveloped maxilla which is responsible for the dental malocclusion in an ogival palate. Other common facial features are palpebral ptosis, strabismus, fine upper lip with drooping commissures, epicanthus, and low implantation of the auricular pavilions. One of the most typical findings in the neonatal period is the presence of lymphedema, a consequence of an alteration in the lymphatic drainage. This anomalous lymphatic drainage affects the formation of other organs, leaving other sequelae such as pterygium colli or winged neck that comes from the nuchal hygroma present during fetal life: the reabsorption of the same results in the characteristic skin folds of the neck as well as the low implantation of the hair and the detachment of the auricular pavilions. A characteristic feature is the widening of the thorax and shoulders sometimes associated with pectus excavatum. The short thorax and the widening result in an increase in the intermamillary distance (teletelia). The short stature is an almost constant sign, other characteristics of this syndrome are intrauterine growth retardation, slow growth from early childhood, and absence of the outbreak of pubertal growth with delayed bone maturation.
TS is associated with other pathologies or diseases, cardiovascular disorders are the most frequent and are usually present in up to 50% of patients. The most frequent congenital heart diseases are aortic coarctation and bivalve aorta. The main causes of prenatal mortality secondary to congenital heart defects are hypoplastic left heart syndrome and/or hypoplastic aorta1. There may also be renal alterations and autoimmune diseases2,3.
The anteromedial diaphragmatic defect through the Morgagni hole "Hernia de Morgagni" represents 2% of the diaphragmatic hernias. Defect produced by the lack of fusion of the sternal and crural portion of the diaphragm, being more frequent that of the right side (90%), they can be bilateral. The hernia sac usually contains the transverse colon, the small intestine, or the liver. The majority of children with this defect are asymptomatic and are diagnosed after the neonatal period, the defect is detected incidentally or is identified as part of the diagnostic approach of respiratory symptoms (tachypnea) and intestinal symptoms such as abdominal pain, vomiting, or constipation. Chest radiography is usually the study in which the defect is identified during the neonatal period, where a structure is identified behind the cardiac silhouette and the lateral projection allows this structure to be located in the retrosternal area. There may be other alterations or syndromes associated with Morgagni's diaphragmatic defect, among which are intestinal malrotation, esophageal atresia, omphalocele, trisomy 21, 18, and 13, Beckwith-Wiedemann syndrome, and Goldenhar syndrome5,6.
Clinical case
Female newborn product of the second gestation; mother of 19 years old, regular prenatal control, at 14 weeks of gestation the prenatal ultrasound reports hydrops fetalis, continuing prenatal follow-up in the gynecology-obstetrics service of the General Hospital of Mexico "Dr. Eduardo Liceaga" O.D., where amniocentesis and amniotic fluid karyotype is performed, reporting a 45X chromosomal complement (Fig. 1), later enrolled with normoevolutive pregnancy. It is born vaginally, meconium amniotic fluid ++, and non-vigorous product, it is aspirated by air, and a ventilation cycle with positive pressure is applied, Apgar 6/7, weight at birth 3000 g, height 49 cm, and gestational age by Capurro (37.6).

Figure 1 Karyotype from amniotic fluid. 30 metaphases were analyzed with GTG banding technique and chromosomal resolution from 375 to 400 bands. A 45X chromosomal complement was found.
The physical examination revealed dolichocephalic skull, anterior fontanelle of 2 cm × 1 cm, telecanthus, palpebral ptosis, low implantation of auricular pavilions, micrognathia, pterygium colli (Fig. 2), enlarged thorax, teletelia (Fig. 3), heart sounds and pulmonary fields without alterations, full limbs, lymphedema of hands and feet, adequate muscle tone, anus, and esophagus watertight; the rest of the physical examination without alterations.
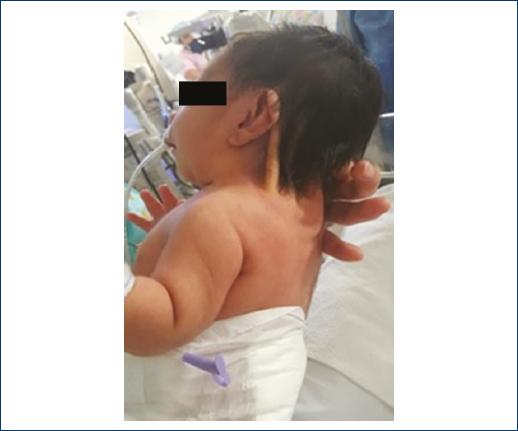

At 10 min of age, she started with respiratory distress, Silverman-Andersen of 2 (mild nasal flaring and discrete intercostal drainage) so that she was admitted to the neonatal intermediate therapy service, where the following study approach was performed: support with supplementary oxygen with a cephalic helmet with FiO2 28-40%, thoracoabdominal radiography is performed (Fig. 4) for persisting with tachypnea, where opacity is observed in the right hemithorax basal area, so angiography of the thorax is performed (Fig. 5) that reports images related to pleuropericardial cyst. An echocardiogram is performed that reports: 5 mm × 2 mm × 1 mm patent ductus arteriosus without hemodynamic repercussion, pulmonary normotension with PSAP 19 mmHg, and ventricular function conserved with 65% FEVi. The service of pediatric surgery is evaluated by those who perform thoracotomy with diaphragmatic plasty with Morgagni Hernia with a 4 cm × 3 cm defect, no complications are made during the procedure; it is possible to remove the support with supplemental oxygen 8 days after surgery. It is discharged 20 days after the surgical procedure, without the need for supplemental oxygen and in adequate conditions.


Discussion
TS was described in 1938 by Henry Turner. In 1958, Ford found that these patients had 45 chromosomes, with a single X chromosome. TS is one of the most frequent human chromosomal disorders, affects one girl for every 2500 newborns alive. As fetal chromosomal anomaly is even higher frequency, 99% of pregnancies with fetus 45 X end in spontaneous abortion, a fact that occurs mainly in the first trimester so that only those fetuses with "moderate forms" syndrome Turner are viable7.
The phenotypic expression of ST can be explained based on three theories: (1) state of haploinsufficiency of genes that are normally expressed in the two sex chromosomes and that would escape the phenomenon of inactivation, (2) imprinting phenomenon that modifies the expression of the gene depending on their paternal or maternal origin, and (3) non-specific defects secondary to chromosomal imbalance. The wide spectrum of somatic signs present in the ST indicates that different genes located on the X chromosome may be responsible for the complete phenotype3,7.
Cases diagnosed with ST are usually due to incidental findings such as pterigium colli, hidrops fetalys by prenatal ultrasound from 11 at the 13.6 weeks of gestational age, and ocasionally the abnormal levels of biochemical markers of the first trimester, such as the fraction of human chorionic gonadotropin (β), alpha-fetoprotein and the evidence of the cytogenetic study in villus biopsy corrals or in amniotic fluid and currently with the study of fetal DNA in maternal plasma that provides definitive information3. Genetic counseling must be carried out in a multidisciplinary manner with the participation of the endocrinologist pediatrician who is the one who knows best the long-term evolution and all aspects related to the prognosis and quality of life of these patients4.
The TS is a well-defined clinical entity that in recent years has experienced considerable progress in terms of knowledge of its pathophysiology and the treatment of comorbidities. The scientific knowledge has allowed to change some aspects such as the realization of an early diagnosis, timely treatment of short stature, treatment of hypogonadism, possibility of pregnancies through assisted reproduction programs, psychological support, social help through the groups of support and a better treatment of the associated pathology, greater life expectancy, better quality of life, and satisfactory incorporation into the social and labor world1. The mortality associated with TS is 3 times higher than in the normal population and the life expectancy is reduced in 13 years, especially in 45X women, with cardiovascular disease being the main cause of death. A multidisciplinary team is needed that also includes the endocrinologist pediatrician, geneticist, cardiologist, nephrologist, otolaryngologist, surgeon, ophthalmologist, gynecologist, psychologist, and orthopedist able to assume the diagnosis and treatment of all comorbidities2,3.
Historically, congenital diaphragmatic hernia (CDH) was considered a surgical emergency; newborns were immediately taken to the operating room as soon as possible with the belief that reducing the abdominal contents of the chest would relieve lung compression. However, after a short post-operative period, they presented respiratory deterioration, high pulmonary vascular resistance, right-to-left shunting, hypoxemia, and death in the final period resulting from respiratory failure. Therefore, a period of medical stabilization and delay in repair was proposed in an attempt to improve the general condition of the newborn with HDC. The survival rate of newborns with CDH ranges from 60 to 90% through the use of physiological treatment strategies including gentle ventilation techniques, high-frequency ventilation, cardiovascular pharmacological support, and extracorporeal membrane oxygenation (ECMO) with gradual improvement in survival5,6.
ST has been described in association with other disorders such as autoimmune diseases including inflammatory bowel disease, rheumatoid arthritis, autoimmune thyroiditis, diabetes mellitus, and, in rare cases, coexisting with Klippel-Feil syndrome8; however, after the extensive review of literature, there are no reports of diaphragmatic defects with TS, reason for the importance of the presentation of this clinical case.

