











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Hereditary angioedema (HAE) is a genetic disease characterized by the presence of transitory and recurrent submucous and/or subcutaneous edema that results in abdominal pain and/or swelling.1 It is classified as a rare disease due to its low incidence (approximately 1-9 cases in every 100,000 individuals) with a great impact on quality of life and life-threatening in some cases.2
There are three types of HAE; Type I (85 % of cases) is caused by a deficit in C1 inhibitor.3,4 This C1 esterase inhibitor controls C1 protein from the complement system C1-C9 family members. Low levels of a C1 inhibitor and C4 suggest HAE due to C1 deficit.5 Type II HAE (15 % of cases) is due to a functional deficit of C1 inhibitor with normal or elevated levels of the protein.3,4
Type I and II HAE are life-threatening conditions with autosomal dominant inheritance pattern characterized by mutations in SERPING 1 gene, where 20-25% are de novo mutations with negative family history. Bradykinin-mediated angioedema is the main cause of HAE with C1 inhibitor deficiency.3,4 Instead, acquired angioedema develops mainly due to an underlying disease characterized by increased consumption of C1 inhibitors and/or development of autoantibodies against C1 inhibitor protein.5,6
Type III HAE resembles types I and II, but with normal C1-Inhibitor determination and estimated prevalence of 1 in 40 000 individuals.7,8,9 Six mutations have been identified: four with an autosomal dominant pattern (coagulation factor XII gene, ANGPT1, KNG1 and MYOF-217s) and two with nonsense mutations (kringle 3 and HS3ST6). Type III HAE patients lacking mutations are classified as unknown (UNK-HAE).10,11,12,13
Mexico lacks epidemiologic data on HAE, generating uncertainty about presentation characteristics of the disease. Several reports indicate HAE underdiagnosis due to general practitioners’ unfamiliarity with disease’s symptoms, confusing it with other diseases. Consequently, a correct diagnosis can be delayed for years.14,15
Because HAE is a rare disease, information gathering on patient journey and effects on quality of life can be complicated. The aim of this study was to perform a survey on HAE patients to understand their journey regarding symptom initiation, route to diagnosis, treatment allocation and follow-up as well as to evaluate burden of disease from a patient’s perspective.
Methods
A cross-sectional survey was administered to Mexican HAE patients between June 15th and September 20th, 2022. It contained 127 questions divided in five sections: Section 1 covers patient’s journey from the moment symptoms started to the moment of diagnosis, treatment allocation, disease evolution, follow-up by health professionals and accompaniment by caregivers. It was built by the authors, based on institutional clinical practice guidelines.16,17,18,19,20 Section 2 details disease impact on daily life activities such as work productivity, daily activities and academic performance. 21 Section 3 evaluates quality of life deterioration through the questionnaire AE-QoL, based on four domains: mobility, fatigue/mood, fear/shame and nutrition.22 Section 4 evaluates the presence and severity of depressive symptoms using the Hamilton depression scale (HADS).23 Section 5 evaluates presence and severity of anxiety symptoms using the Hamilton anxiety scale.24
Patients were recruited through patient organization named “AHSI Luchemos Por La Vida” or through private practice. The main researcher explained purpose and procedures of the study. Patients included clinically stable adults diagnosed with HAE who agreed to participate by 1) signing an informed consent form and
2) could fill out the survey by themselves or aided by a trusted caregiver. The study was authorized by Research Committee (CI-000002), Research Ethics Committee (CEI-000002) and registered under CONBIOETICA.
Descriptive analysis for qualitative variables consisted of frequency and percentages, while quantitative variables are presented with mean (SD) and median (IQR) depending on data dispersion. All statistical analyses were performed in Excel 2016.
Results
Patient Characteristics
17 patients who responded completely or partially were included for analysis. Demographic characteristics are shown in Table 1. Two patients were male (11.8%) and 88.2% were female (n = 15), with ages ranging from 23 to 67 years. 8 patients (47%) were treated at ISSSTE, 7 at IMSS (41.2%) and 11.8% attended their local Health Ministry clinics. 12 patients (70.6%) had at least one relative diagnosed with HAE, 76.5% were married (n = 13) and 14 had children, 6 of whom had at least one child diagnosed with HAE.
Patient journey
The most common clinical presentation of HAE was Type I, with 70.6% of cases (n = 12), followed by normal C1 inhibitor HAE in 11.8% (n = 2). 17.6% of patients (n= 3) were not aware of the type of HAE diagnosed.
Mean age at diagnosis was 36 (median= 33) years (Figure 1), while average time between symptom onset and diagnosis was 20 (median = 22) years.
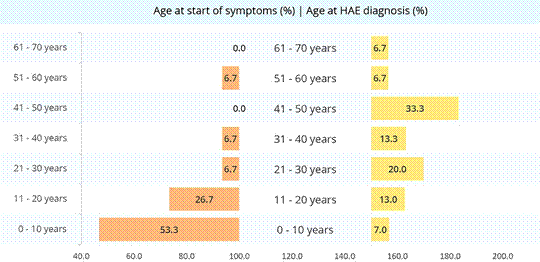
Average age at start of symptoms was 16 (median = 10) years. By age 10, 53% of patients (n = 8) had experienced some type of symptom, with the earliest presentation beginning at 2 years old. 26.7% had their first symptoms between ages 11-20 and 20% after turning 20, with maximum starting age at 53 years, and 13.3% of patients did not refer age of symptoms onset.
Waiting time for specialist evaluation in public sector ranged from 10 days to 32 years, and zero for private sector. Specialists tend to request laboratory tests upon first visit, including complement levels, urine analysis and blood levels of C1 inhibitor and C4.
Time elapsed between symptoms onset and first consultation ranged from zero days to 10 years, with 41.2% of patients (n = 7) claiming they received care at first level of care on the day symptoms started. On average, they visited nine different physicians before reaching definitive HAE diagnosis, with 88.2% of patients referring they received three different diagnoses before HAE, and twelve (80%) received an allergy related diagnosis. Patients with incorrect diagnosis received treatment for at least one month, and up to 30 years (Figure 2). These patients had at least one episode a year and up to 260 episodes before receiving a HAE diagnosis, with an average of 5 episodes a month. In 94.1% of cases, they referred several affected areas, the most frequent being the face 71% (n = 12), abdomen 71% (n = 12), hands 76% (n = 13), arms 53% (n = 9), feet 53% (n = 9), larynx 47% (n = 8), legs 41% (n = 7), and genitalia 41% (n = 7). One patient referred a single area (abdomen), and other referred a full body episode (except the ears).

Regarding HAE treatments, 58.8% of patients (n = 10) could identify up to 2 treatments, 17.6% (n = 3) could name 3 to 6 and 11.8% (n = 2) were not aware of any. A total of 64.7% patients (n = 11) had access to treatment.
Initial treatments included danazol, nadroparin, fresh frozen plasma, enoxaparin, montelukast, chloropyramine, loratadine, celecoxib, fexofenadine, dexamethasone, and hydrocortisone. Two patients mentioned no initial treatment. Currently, patients are treated with enoxaparin, icatibant, nadroparin, loratadine, danazol, fexofenadine, fresh frozen plasma and five patients mentioned no current management. Only 64.7% (n =11) considered their treatment effective.
Four patients out of 17 who answered the question “Did you receive prophylactic treatment?”, mentioned receiving long term preventive treatment as efficient, as they ease swelling and stop or prevent crisis. On average, patients changed treatment in three occasions. Causes cited for treatment abandonment included treatment unavailability from provider, problems administering treatment, treatment discontinuation, complicated bureaucratic procedures, and side effects.
Five patients required psychological support (29.5%), two (11.8%) required occupational therapy and four (23.5%) received other treatments such as analgesics, sedatives, alternative medicine, and psychiatric evaluations. 52.9% of patients (n= 9) required frozen fresh plasma, 17.6 % (n= 3) received adrenaline and 3 patients required ophthalmologic evaluation. Respiratory support, endoscopy and surgery was required by 2 patients (11.8%) and one patient required tracheostomy. 88.2% of patients referred recurrent episodes of angioedema, caused by physical strain, footwear changes, heat, stress, lack of sleep or no identifiable cause.
All patients referred some impact of HAE on their social life by avoiding activities they would normally enjoy depression due to social distancing, fear, anger, anxiety, and a decreased quality of life that prevents them from leading a “normal” life, problems making shortor long-term plans and trips, as well as a strong impact at the knowledge of the hereditary factor of their disease, including problems with their significant others.
Patients refer conflict with the nature and diagnosis process of their disease, questioning the lack of diagnosis that caused deaths within the family and contemplating their own mortality.
The evolution time since HAE diagnosis was 13.7 years (SD 6.9), with 35.3% of patients referring the need of a caregiver. Care was provided by relatives without economic remuneration. The evolution time since start of symptoms was 32.3 years (SD 15.1) with a median of 30 years.
Work, academic performance and daily life activities affectation.
Seven patients (41.2%) were unemployed (retired, homemakers and students). 52.9% of patients had a gainful employment (n= 9) with an average of 33.3 work hours a week and referring an average loss of 9.7 work hours due to HAE. Two patients were students and had classes 17 and 42 hours a week, one of them referred missing 32 hours a week to large detriment of his academic performance, the other student referred no missing time or negative effect. 64.7% of patients mentioned some negative effects on their daily life activities, 17.6% referred no effects and three patients did not answer.
Quality of life
Twelve patients (70.5%) referred at least some kind of negative effect on their physical activity, 11 (64.8%) had some effect on spare time and social relationships were negatively impacted in12 patients (70.5%), see Figure 3. Eating habits and nutrition were affected in some degree in 53% of cases (n= 9), whereas 14 patients (82.3%) reported some level of fatigue.
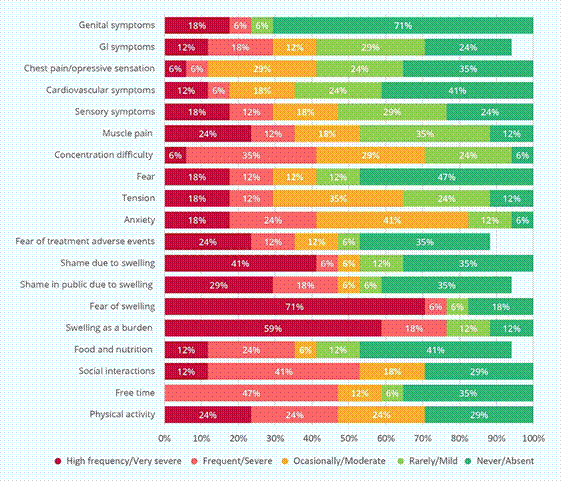
Nearly 60% of patients (n = 10) mentioned swelling as a very frequent burden, 70.6% cited fear of swelling and 58.8% (n = 10) feel shame to go out in public due to swelling to different degrees. Furthermore, almost two thirds of patients mentioned some degree of fear to treatment side effects.
Anxiety
Very severe anxiety and tension were present in 17.6% each, and more than half of the participants (52.9%, n= 9) had some level of fear related to their condition. The 82.3% referred some level of insomnia and 94.1% had some level of concentration difficulty; 88.2% presented some level of sensory symptoms (ear buzzing, blurry vision, chills, tingling, etc.). Cardiovascular and gastrointestinal symptoms were present on 58.8% and 76.5% of patients, respectively. See figure 3 for detailed severity of symptoms distribution.
Depression
Seven patients indicated some expression of depression (41.2%). One patient disclosed two suicide attempts before HAE diagnosis, and another expressed suicidal ideation due to the disease mentioning “life is not worth living with HAE”. 35% of patients had some communication issues, ten patients presented some level of somatic anxiety (59%), 29.4% had some level of appetite loss and a similar percentage had some degree of active symptoms. 41.2% of patients were aware of their condition, although they do attribute it to their own habits, workload, virus infections or others.
Discussion
This is the first study providing insights into the patient journey of Mexican patients with HAE regarding risk factors, quality of life affectation, time taken to care, correct diagnosis, and treatment. It would help to create awareness in governmental and health sector, supporting decision making when designing and prioritizing interventions tailored for optimization in control and management of the disease by adopting patient-centered strategies, according to Devi and collaborators.25
The autosomal dominant hereditary pattern of the disease was evident in this work, where 70.6% of patients had family members diagnosed with HAE and 35% had children diagnosed as well. Our results show that age of presentation had a wide range, starting at 2 years old and up to 53, consistent with other reports.19,20 This work confirms Mendez and Quiroga’s finding that diagnosis is commonly performed late (second and third decade of life), especially in cases without family history and even when symptoms start in childhood or adolescence.26
We found a delay in diagnosis up to 42 years, with shorter timespans in patients who had family history of HAE and those who seek out a specialist for diagnosis. However, when there’s no family history or first contact is established through a general practitioner and/or first contact clinic environments, the diagnosis takes longer and symptoms tend to be adjudicated to other more common conditions, mainly allergies.20
Our findings coincide with previous reports that HAE has a significant negative impact on patient’s quality of life due to the recurrent and unpredictable nature of the episodes that result in limitations of their social lives, travels, and physical activity, with more severe episodes requiring urgent intervention, visits to the emergency room or hospitalization.22 In patients where affected areas included throat, larynx and trachea, the episodes can be life-threatening without proper treatment.27 One patient mentioned some of them can die before receiving an accurate diagnosis, for a disease that even if there is no cure, adequate treatment and follow-up can improve quality of life and increase life expectancy.1
However, despite medical advances for HAE during the last decade, patients still face significant burden related to disease and its treatment.28 Our research showed that nearly 60 % of patients were aware of a couple of available treatments while only 11 of the 17 participants had access to treatment.
A consensus for HAE in Argentina emitted a recommendation evaluating the need for long-term prophylaxis against episodes in all patients, considering aspects related to episodes, treatment access and the impact of episodes on patient’s quality of life.29 However, our findings show that only four patients were under long-term prophylactic treatment.
One limitation of our study is the lack of validation for the instrument section on patient’s journey. Therefore, results may be biased due to recall bias and question comprehension by the participants. Nevertheless, is the first of its kind and provides valuable information on patient journey for Mexicans with HAE.
Patients living with chronic rare diseases like HAE have higher psychological, social, economic, and cultural vulnerability;1 therefore, anxiety and depression are common amongst them.30 Our study found concurrent evidence of anxiety and depression (41.2% each).
Conclusions
Hereditary Angioedema is a rare disease with huge social impact on patients, as it limits daily living activities including work, travel, social and physical activity amongst others. This worsens a quality of life already diminished by symptoms recurrency, little information on the disease, low awareness of general practitioners for its diagnosis, treatment barriers and depression and anxiety due to their situation.
We need to reinforce knowledge and awareness of first contact and general practitioners about rare diseases to improve early diagnosis of HAE. It is also necessary to increase awareness of decision makers in the public health sector to promote treatment access. Finally, general population must be made aware of the burden of the disease. We hope this work, first of its kind in Mexico, can be a starting point for further research.
Conflict of interests
The VCME author has received consultancy (advisory boards) y and speaker fees from Takeda, Sanofi, Astra Zeneca, CSL Behring, and support research grants Takeda, Sanofi, Astra Zeneca y Chiesi. The authors GVS, GFH y BLYO are responsible for the content of the article, they are employees of HS Estudios Farmacoeconomicos S.A. de C.V., are members of ISPOR and state that they have received fees from AstraZeneca, Biogen, Medix, Merk, Novartis, Pfizer, Roche, Sanofi and Takeda. The authors have no other relevant affil iation or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript other than those disclosed.
Ethical considerations
The present study was carried out in accordance with the ethical principles that originate in the Declaration of Helsinki, with the Good Clinical Practice Guidelines (ICH, Topic E6, 1995), as well as the Regulations of the General Law of Health in Mexico on Health Research. The study was authorized by Research Committee (CI000002), Research Ethics Committee (CEI-000002) and registered under CONBIOETICA.

